СОЕДИНЕНИЯ С КРАТНЫМИ СВЯЗЯМИ МЕТАЛЛ-УГЛЕРОД В КАТАЛИТИЧЕСКИХ РЕАКЦИЯХ МЕТАТЕЗИСА |
|
Нобелевская лекция, 8 Декабря 2005 г. |
|
Ричард Ройс Шрок |
 |
Химический факультет 6-331, Массачусетский Технологический Институт, 77 Массачусетс Авеню, Кембридж, Массачусетс 02139, США. |
|
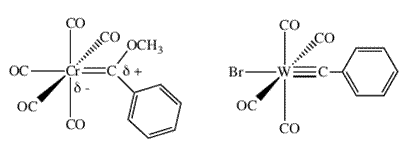
Для меня большая честь находиться сегодня здесь, в состоянии, о котором я бы никогда не подумал, что оно возможно. Я надеюсь, что история, которую я сейчас вам поведаю, породит у вас некоторое представление о том, какой вклад я внес в ту область, за которую в этом году была присуждена Нобелевская премия по химии. Эта история началась тридцать два года тому назад в 1973, в этот год Нобелевская премия была поделена между Г. Вилкинсоном и Э.О. Фишером. Нобелевская лекция Вилкинсона 1 касалась природы простой связи между переходным металлом и атомом углерода в алкильной группе, и в ней подчеркивался тот факт, что связь металл-углерод по своей природе не является слабой. Э. O. Фишер в своей Нобелевской лекции 2 обобщил обширные сведения о химии «карбеновых» комплексов переходных металлов 3,4, в которых имеется металл-углеродная двойная связь, открытых им и его группой в 1964 (схема 1) 5. Он также сообщил о новых «карбиновых» комплексах, содержащих металл-углеродную тройную связь 6. Было ясно, что металл-углеродная одинарная связь имеет огромное значение в зарождающейся области гомогенного катализа. Впрочем, каталитические реакции с участием молекул с двойными или тройными металл-углеродными связями, были еще неизвестны. Когда я обратился в Центральный исследовательский отдел E. I. DuPont de Nemours and Company в 1972, органическая химия переходных металлов и гомогенный катализ вызывали большой интерес по причине их огромного потенциала в органической химии, а следовательно и в промышленности. В начале 1970-х химики-неорганики знали, что многие молекулы комплексов переходных металлов, содержащих металл-углеродную связь, вступают в различные реакции разложения, которые являются значительно более быстрыми, чем молекулы непереходных металлов, таких как Zn(CH2CH3)2 или Al(CH2CH3)3. Наиболее обычной из их числа заключалась в переносе 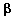 -водородного атома от этильной группы (MCH2CH3), например, к металлу с образованием гидрида металла и алкена. -водородного атома от этильной группы (MCH2CH3), например, к металлу с образованием гидрида металла и алкена. |
|
 |
|
| Схема 1. Карбеновые (слева) и карбиновые комплексы (справа) с металлами в состоянии с «низкой степенью окисления». | |
 |
|
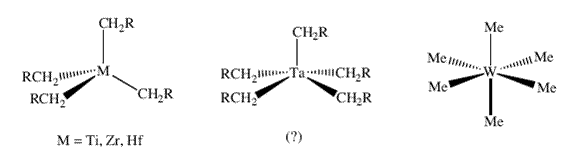
| Схема 2. «Пералкильные» комплексы металлов 4, 5 и 6 групп в состоянии с максимально возможной степенью окисления. | |
Относительная стабильность «гомолептических» или «пералкильных» соединений металлов в высокой степени окисления, таких как M[CH2Si(CH3)3]4, M(CH2C6H5)4 и M[CH2C(CH3)3]4 (M = Ti, Zr, или Hf; схема 2), была объяснена на основании того факта, что в отличие от соединений с этильным лигандом, алкильные лиганды в этих молекулах не имеют -водородных атомов и поэтому, естественно, не могут вступать в процессы разложения с участием -атомов водорода 7. В 1973 Вилкинсон опубликовал статью с описанием синтеза W(CH3)6 8. В отличие от молекулы M(CH3)4 (где M = Ti, Zr или Hf) W(CH3)6 стабилен при 22°C. Метильный атом углерода является 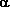 -атомом по отношению к атому металла; в данном случае -углеродные атомы отсутствуют, как, соответственно и -атомы водорода. Однако метильные производные не защищены стерически от бимолекулярных реакций с участием -водородных атомов. Как можно увидеть из примечания, добавленного к ссылке 9 на речь Вилкинсона по поводу вручения ему Нобелевской премии , я был заинтригован высокой степенью окисления металлов в пералкильных комплексах и сделал свой выбор в пользу изучения химии металлорганических соединений тантала вскоре после моего прибытия в DuPont. В то время о химии алкильных производных металлов 5 группы (V, Nb, Ta) было известно немного; я выбрал тантал потому, что он является ближайшим соседом вольфрама, находящегося в 6 группе (Cr, Mo, W), и подобно вольфраму, тантал относительно стабилен в состоянии с наивысшей возможной степенью окисления, Ta(5+). Поэтому пералкильными комплексами тантала должны быть пентаалкилы. Стартовой точкой для химии алклильных комплексов тантала послужила статья Г.Л. Жувиналя 10, в которой он описал получение с низким выходом триметилдихлоридов ниобия и тантала путем прибавления диметилцинка к пентахлоридам металлов. Я обнаружил, что кристаллический TaMe3Cl2 не только можно получать с количественным выходом в пентане в больших количествах (схема 1), но и ио, что его можно длительное время хранить при – 40 °C в твердом состоянии. -атомом по отношению к атому металла; в данном случае -углеродные атомы отсутствуют, как, соответственно и -атомы водорода. Однако метильные производные не защищены стерически от бимолекулярных реакций с участием -водородных атомов. Как можно увидеть из примечания, добавленного к ссылке 9 на речь Вилкинсона по поводу вручения ему Нобелевской премии , я был заинтригован высокой степенью окисления металлов в пералкильных комплексах и сделал свой выбор в пользу изучения химии металлорганических соединений тантала вскоре после моего прибытия в DuPont. В то время о химии алкильных производных металлов 5 группы (V, Nb, Ta) было известно немного; я выбрал тантал потому, что он является ближайшим соседом вольфрама, находящегося в 6 группе (Cr, Mo, W), и подобно вольфраму, тантал относительно стабилен в состоянии с наивысшей возможной степенью окисления, Ta(5+). Поэтому пералкильными комплексами тантала должны быть пентаалкилы. Стартовой точкой для химии алклильных комплексов тантала послужила статья Г.Л. Жувиналя 10, в которой он описал получение с низким выходом триметилдихлоридов ниобия и тантала путем прибавления диметилцинка к пентахлоридам металлов. Я обнаружил, что кристаллический TaMe3Cl2 не только можно получать с количественным выходом в пентане в больших количествах (схема 1), но и ио, что его можно длительное время хранить при – 40 °C в твердом состоянии. |
|
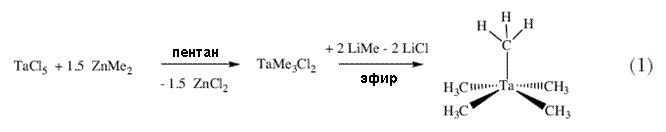 |
|
Более того, он реагирует с двумя эквивалентами LiMe с образованием летучего, желтого кристаллического TaMe5 11. Это вещество значительно менее стабильно, чем W(CH3)6, однако намного более устойчиво, чем Hf(CH3)4. Пентаметилтантал разлагается при температуре выше 0 °C с образованием ~3.7 эквивалентов метана, и происходит это в бимолекулярной реакции. В пентаметилтантале имеются 15 стерически незащищенных-C-H связей и металл является в высокой степени электронодефицитным (10 электронов, ему не хватает 8 электронов до заветного числа электронов 18). Тем самым оказывается содействие взаимодействиям между металлическим центром и CH связями другой молекулы комплекса, они протекают легче в TaMe5, чем в WMe6 по простым стерическим соображениям. |
|
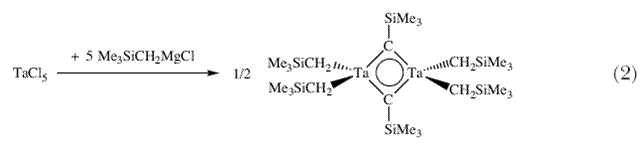 |
|
Вилкинсон недавно опубликовал работу с описанием реакции, которая привела к образованию необычной димерной молекулы, содержащей по его словам «карбеновый мостик»  -CSiMe3 (фактически карбиновый мостик; схема 2). В отношении Ta(CH2SiMe3)5 он сказал «Считается, что пентаалкильный комплекс не может существовать по стерическим причинам» 12. Поэтому я обратился к экспериментам с неопентильным лигандом (CH2CMe3) с целью определить предел стерических препятствий в гомолептических пентаалкильных соединениях тантала d0 и природу путей их разложения, что можно было бы определить при этих обстоятельствах. Ключевой эксперимент (схема 3) заключался в попытке получения Ta(CH2CMe3)5 путем прибавления двух эквивалентов LiCH2CMe3 к Ta(CH2CMe3)3Cl2. -CSiMe3 (фактически карбиновый мостик; схема 2). В отношении Ta(CH2SiMe3)5 он сказал «Считается, что пентаалкильный комплекс не может существовать по стерическим причинам» 12. Поэтому я обратился к экспериментам с неопентильным лигандом (CH2CMe3) с целью определить предел стерических препятствий в гомолептических пентаалкильных соединениях тантала d0 и природу путей их разложения, что можно было бы определить при этих обстоятельствах. Ключевой эксперимент (схема 3) заключался в попытке получения Ta(CH2CMe3)5 путем прибавления двух эквивалентов LiCH2CMe3 к Ta(CH2CMe3)3Cl2. |
|
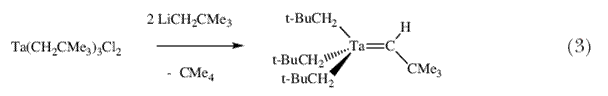 |
|
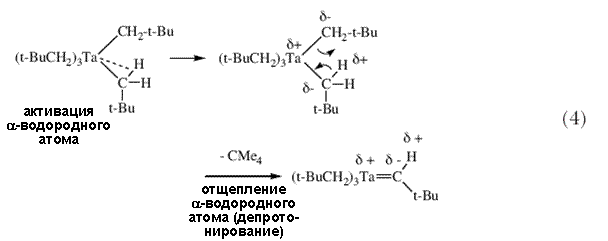
| Вместо Ta(CH2CMe3)5 с количественным выходом образовался оранжевый кристаллический и термически стабильный (Me3CCH2)3Ta=CHCMe3 13. Точный механизм этой реакции пока неизвестен, однако вполне вероятно, что -водород активируется атомом металла и впоследствии может быть отщеплен неопентильной группой, вероятно в результате внутримолекулярной реакции самого пентанеопентилтантала (схема 4). |
|
 |
|
(Me3SiCH2)3Ta=CHSiMe3 может быть интермедиатом в реакции, показанной на схеме 2, однако он должен разложиться внутримолекулярно с образованием тетраметилсилана и наблюдаемой димерной молекулы; в случае (Me3CCH2)3Ta=CHCMe3 этого не наблюдается. Неопентилиденовый комплекс тантала, (Me3CCH)3Ta=CHCMe3, был новым во многих отношениях. Это был первый пример стабильного соединения переходного металла с фрагментом M=CHR. Во-вторых, терминальная алкилиденовая группа образовалась в результате нового типа реакции, внутримолекулярного отщепления |
|
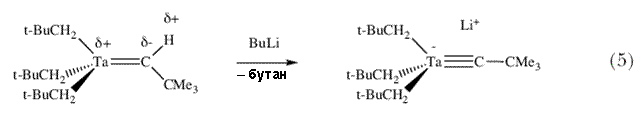 |
|
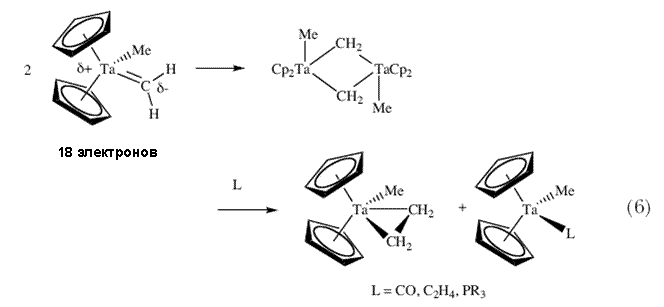
И наконец, было показано, что даже метиленовые (Ta=CH2) комплексы могут быть получены в результате депротонирования [TaCp2(CH3)2]+ 15,16. Даже если TaCp2(CH3)(CH2) содержит 18 электронов на внешних орбиталях металла, это вещество является нестабильным в отношении бимолекулярного разложения с образованием комплекса с этиленом с выходом 50%, и родственных молекул, которые образуются в присутствии некоторых лигандов L (схема 6). Реакции бимолекулярного разложения алкилиденов, в особенности метиленов, являются предметом споров, которые продолжаются по сей день. |
|
 |
|
| Основным достижением между 1973 и 1975 было получение метиленовых и метиновых комплексов тантала с высокой степенью окисления металла, и они оказались по некоторым важным критериям существенно отличными от комплексов, полученных Фишером. В определенный момент времени в начале 1970-х мне стало известно о реакции «метатезиса олефинов» (схема 7), поразительной и загадочной реакции, катализируемой гомогенными катализаторами на основе Mo и W (и протекающей также на гетерогенном Re-катализаторе), природа которых была неизвестна 17-20. Физико-органические эксперименты, проведенные (в числе других) Р.Х. Грубсом 21-23, Т.Д. Катцем 24,25, К.П. Кэйси 26, и Д. Шовеном 27, были специально разработаны с целью определения того, что реакция протекает по «парному» или «непарному» механизму 20. | |
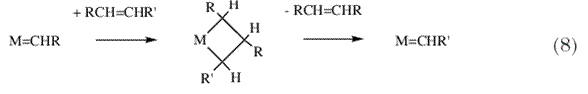 |
|
| Было показано, что реакция протекает по «непарной » схеме, и Шовен был первым, кто предложил верный «карбен/металлоциклобутановый» механизм реакции (схема 8) , который объяснял все наблюдаемые факты 27. Однако в то время не были известны «карбеновые» комплексы, которые бы эффективно реагировали с обычными олефинами, как это предположил Шовен. Также был обнаружен метатезис алкинов (схема 9), как гетерогенный 28,29 так и гомогенный 30, катализируемый соединениями Mo и W неизвестного строения,. Т.Д. Катц 24 предположил механизм метатезиса алкинов, аналогичный механизму метатезиса алкенов (схема 10), т.е. механизм, включающий в себя образование интермедиатов металлоциклобутанового строения. В то время пока еще были неизвестны «карбиновые» комплексы, которые бы катализировали реакции метатезиса алкинов. | |
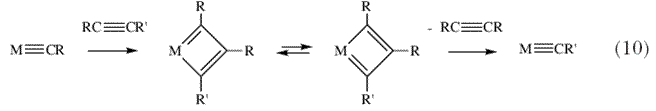 |
|
| Я спросил себя, а не могут ли эти новые «алкилиденовые» и «алкилидиновые» комплексы тантала быть по крайней мере тем типом молекул, которые способны катализировать реакции метатезиса олефинов и ацетиленов, соответственно, даже если в настоящее время ничего не известно о том что тантал может выступать в качестве катализатора каждой из этих реакций. После моего переезда в MIT в 1975 г., я начал подробно исследовать реакции между различными алкилиденовыми комплексами и олефинами. Выяснилось, что (схема 11) электронодефицитные алкилиденовые комплексы тантала и молибдена легко реагируют с олефинами с образованием металлоциклобутановых интермедиатов, однако эти частицы перегруппировываются в результате -гидридного процесса с образованием всех четырех возможных продуктов перегруппировки вместо потери молекулы олефина и превращения в новый алкилиденовый комплекс (схемы 12 и13); ни в одном из случаев цепная алкилиденовая реакция не запускалась и поэтому образование продуктов метатезиса не наблюдалось 31. |
|
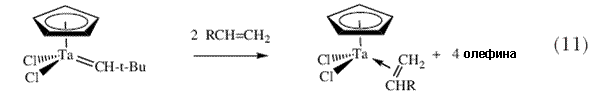 |
|
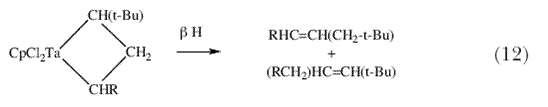 |
|
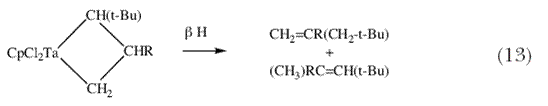 |
|
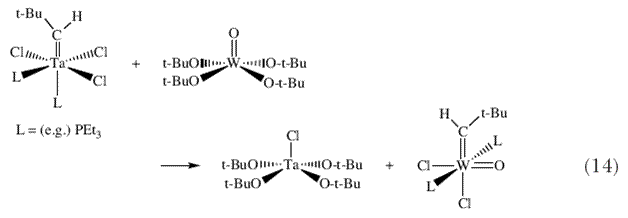
| На Международном симпозиуме по метатезису олефинов (ISOM III), состоявшемся в 1979 г. в Лионе, я доложил о том, что комплексы ниобия и тантала, содержащие трет-бутоксильные лиганды вместо хлоридных лигандов, таких как M(CH-t-Bu)(O-t-Bu)2Cl(PMe3), могут быть вовлечены в реакции метатезиса олефинов. Несколько дюжин каталитических циклов метатезиса можно наблюдать для такого олефина как цис-2-пентен 32. Было высказано предположение, что трет-бутоксидные лиганды «предотвращают восстановление» металла и «способствуют протеканию реакции метатезиса». На той же конференции я сообщил о том, что попытка провести реакцию «подобную реакции Виттига» между алкилиденовым комплексом тантала и оксокомплексом вольфрама с образованием W(CH-t-Bu)(O-t-Bu)4 и Ta(O)L2Cl3, вместо этого неожиданно дала 18-электронный оксо-неопентилиденовый комплекс вольфрама (схема 14) 33. Этот неопентилиденовый комплекс реагирует с олефинами с образованием продуктов метатезиса и новых алкилиденов, образования которых в реакции метатезиса можно было ожидать, включая метиленовый комплекс, и особенно дегко в присутствии следов AlCl3. | |
 |
|
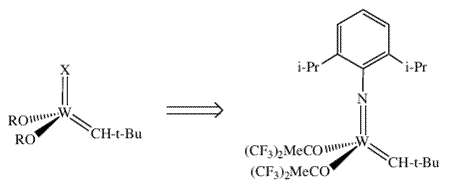
| Это было убедительной демонстрацией того что оксо-алкидиденовые молекулы легко реагируют с олефинами и превращают алкилиденовый комплекс того же типа из половины того количества олефина, которое присутствовало в реакционной смеси. Роль AlCl3, как предполагается, заключается в удалении либо хлора, либо фосфина (обратимо) и тем самым в создании пустого координационного места у электронно насыщенного металлического центра. Считается, что четырехкоординированные неопентилиденовые молекулы, содержащие большие, ковалентно связанные лиганды являются наиболее вероятными выделяемыми, но реакционноспособными электронодефицитными соединениями. Несомненно, неопентилиденовый лиганд был необходим. Мы знали также, что весьма желательным было присутствие алкоксидов, однако что должен был представлять из себя четвертый, и обязательно дианионный, лиганд X (схема 3)? Оксо лиганд был непригоден, поскольку он практически обязательно будет способствовать протеканию реакций бимолекулярного разложения. Мы сфокусировались на имидном ([NR]2-) лиганде, изоэлектронном аналоге оксо лиганда, поскольку имидо лиганд может быть стерически защищен большой группой R. После рассмотрения возможных синтетических трудностей, связанных с тем, что R должна быть группой 2,6-ди-трет-бутилфенильной, мы остановились на 2,6-диизопропилфенильной группе, выбор, который отчасти был ответом на замечание К.Б. Шарплесса, касающегося значения изопропильных групп в целом в сравнении с трет-бутильными группами. | |
 |
|
| Схема 3. Строение стабильного имидоалкилиденбисалкоксидного комплекса вольфрама. | |
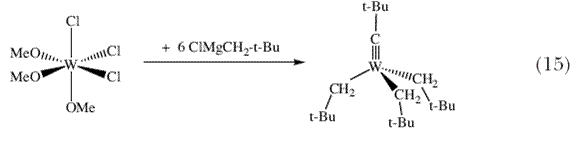
(2,6-диизопропилфенильная группа содержится во многих азот-содержащих лигандах большого числа современных катализаторов). Были доступны различные объемистые и стерически затрудненные третичные спирты и фенолы, включая фторированные трет-бутанолы, такие как (CF3)2MeCOH, pKa которых в воде (~9) имеют значительно меньшие значения, чем сам трет-бутанол (~19). Мы чувствовали, что электронооттягивающие свойства гексафтор-трет- бутоксидного лиганда должны значительным образом увеличивать электрофильность металла и тем самым увеличивать скорость реакции алкилиденового комплекса с олефином. Вследствие этого, мы поставили своей целью получение W(NAr)(CHCMe3)[OCMe(CF3)2]2 (схема 3) в ожидании того, что этот 14-электронный комплекс можно будет не только выделить, но и он будет способен катализировать реакцию метатезиса олефинов. Будучи знакомыми с почти магическими свойствами неопентильного лиганда (равно как и неопентилиденового и неопентилидинового лигандов), мы заинтересовались также простыми реакциями между соединениями W(VI) (первоначально WCl6) и неопентиллитием или неопентилмагний хлоридом, и начали исследовать такие реакции в 1977. Мы обнаружили, что в этих реакциях можно выделить летучий, желтый, кристаллический алкилидиновый комплекс 34, и что реакция между W(OMe)3Cl3 и шестью эквивалентами Me3CCH2MgCl в диэтиловом эфире дает эти вещества с выходом ~50% (схема 15) 35. Комплекс (Me3CCH2)3W CCMe3 по своему строению схож с комплексом (Me3CCH)3Ta=CHCMe3 по наличию неопентильных или производных от неопентильного лигандов вокруг атома металла. CCMe3 по своему строению схож с комплексом (Me3CCH)3Ta=CHCMe3 по наличию неопентильных или производных от неопентильного лигандов вокруг атома металла. |
|
 |
|
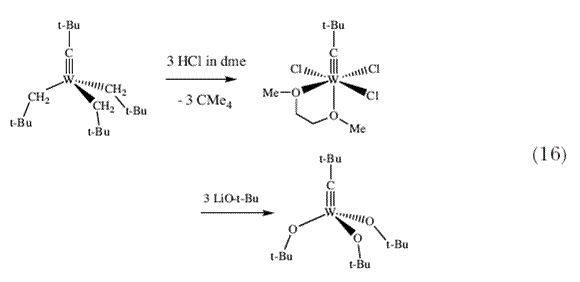
| Когда мы убедились в преимуществах алкоксидных лигандов для эффективного метатезиса олефинов, мы заинтересовались таким вопросом – а нельзя ли получить триалкоксидные молекулы, такие как (Me3CO)3WCCMe3 и не могут ли они служить эффективными инициаторами метатезиса алкинов. С удовлетворением мы обнаружили, что (Me3CO)3WCCMe3 действительно можно легко получить (схема 16), и с особенным удовлетворением обнаружили, что алкины вступают в реакцию метатезиса при комнатной температуре в присутствии комплекса (Me3CO)3WCCMe3 в качестве инициатора 36 со скоростями, в тысячи раз превышающими скорости реакций с катализаторами неизвестного строения, описанными в литературе. |
|
 |
|
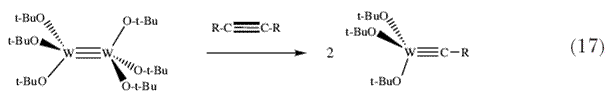
| Эта находка убедила нас в том, что стерически объемные алкоксиды действительно в высокой степени способствуют метатезисной активности. В противоположность (Me3CO)3WCCMe3 не катализирует реакцию метатезиса алкинов, даже если удается выделить и охарактеризовать кристаллографически вольфрамциклобутадиеновый интермедиат, необходимый для протекания этой реакции, например, Cl3W[C(t-Bu)C(CH3)C(CH3)] 37! Наблюдаетя протекание совершенно других реакций 38-40. Комплекс (Me3CCH2)3WCCMe3 также не катализирует реакции метатезиса нетерминальных алкинов; он оказался просто слишком нереакционноспособным. Когда группа OR в комплексе (RO)3WCCMe3 была более электронооттягивающей алкоксидной или феноксидной группой, нам удалось выделить и кристаллографически охарактеризовать промежуточно образующийся фольфрамциклобутадиеновые комплексы в реакциях метатезиса алкинов и подробно изучить их 41,42. В то время были известны многочисленные комплексы, содержащие тройные связи вольфрам-вольфрам или молибден-молибден, молекулы X3MMX3 43,44, и в частности комплекс гекса-трет-бутоксидивольфрам, (Me3CO)3WW(OCMe3)3. Поэтому возникал интересный вопрос, будет ли комплекс (Me3CO)3WCR (схема 17), которые эффективны в реакциях, подобных реакциям метатезиса? |
|
 |
|
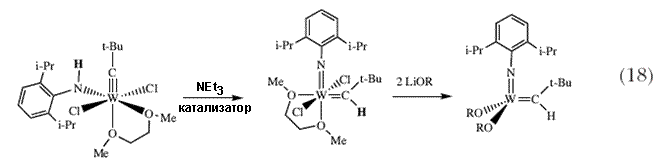
| Ответ оказался положительным, и это происходило на удивление легко 45. Хотя это и не всеобщая реакция для всех комплексов X3MMX3 (M = Mo или W), тот факт, что это является простым только если X является отностительно стерически объемистым алкоксидом, является более чем случайным совпадением. Это открытие открыло путь к соединениям (Me3CO)3WCR, которые не способны к реакциям отщепления -водородного атома 46, и сцементировало взаимоотношение и взаимопревращаемость прочных тройных связей между атомами металла, между атомами углерода (в алкинах) и между атомом металла и атомом углерода в алкилидиновых комплексах. Интересно отметить, что CN связь в нитрилах также легко расщепляется, но в молекуле азота связь NN не расщепляется. Эта удивительная реакция, которая требует (трехкоординированных) триамидных частиц, не публиковалась вплоть до 1995 47. Однако алкилидиновые комплексы вольфрама были важны по другой дополнительной причине. Если неопентилиденовые комплексы могут быть получены путем отщепления -протона от неопентилиденового лиганда, нельзя ли эту реакцию провести в обратном направлении, т.е., нельзя ли присоединить протон к неопентилидиновому комплексу с образованием неопентилиденового комплекса? Кроме того, нельзя ли все это провести таким образом, чтобы параллельно получить другую нужную кратную связь металл-лиганд, а именно имидный лиганд? Амидо/алкилидиновый комплекс фактически можно легко получить, а протон затем перемещается от N к C под действием основания (схема 18). В этом случае легко удается получить большое число комплексов W(NAr)(CH-t-Bu)(OR)2 из дихлорпроизводных, если только OR был достаточно объемистым для того, чтобы противостоять осложнениям, проистекающим от протекания бимолекулярных реакций. |
|
 |
|
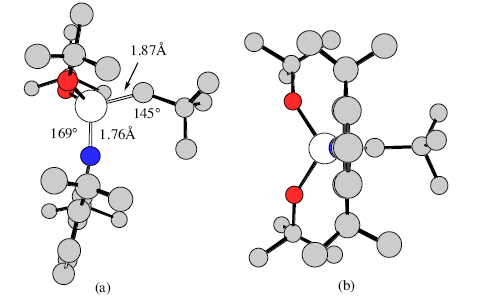
| Кристаллографическая структура, подобная той, что приведена на рисунке 4 показывает, как трет-бутильная группа неопентилиденового лиганда направлена в сторону имидного лиганда (син-ориентация), и как диизопропилфенильная группа у атома азота защищает имидный атом азота и алкилиденовый атом углерода от бимолекулярных реакций. Вскоре стало понятно, что комплексы W(NAr)(CH-t-Bu)(OR)2 ожидаемым образом катализируют метатезис олефинов с активностью, которая в первом приближении коррелирует со способностью лиганда OR оттягивать электроны. Оказалось, что катализаторы, содержащие гексафтор-трет-бутоксидные лиганды, обладают наивысшей активностью. Мы нашли, что вольфрамциклобутановые интермедиаты, т.е. интермедиаты, предложенные Шовеном в реакции метатезиса, действительно могут быть выделены и охарактеризованы кристаллографически 48. Впрочем, в определенных условиях, стабильность вольфрамциклобутанов оказывается проблематичной; в частности, может наблюдаться относительно медленное отщепление олефина от незамещенного вольфрамциклобутанового интермедиата. | |
 |
|
| Рисунок 4. Строение W(NAr)(CH-t-Bu)(O-t-Bu)2; (a) вид сверху; (b) вид сбоку. | |
 |
|
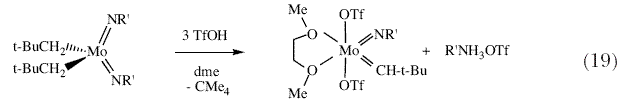
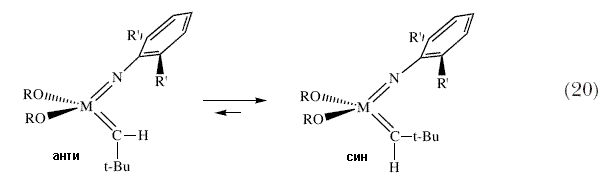
| По этой причине металл остается изолированным в форме вольфрамциклобутана. Поскольку связи Mo-лиганд в общем слабее, чем связи W-лиганд, мы почувствовали, что молибденциклобутановый комплекс может отщеплять олефин более быстро. Поэтому, мы поставили себе задачу синтезировать аналогичные Mo(NAr)(CH-t-Bu)(OR)2 катализаторы. Был разработан синтез бисалкоксидов, ключевой особенностью которого является использование имидо «защитной группы», которая затем удаляется после присоединения трифлата (схема 19), тем самым образуя в ходе процесса неопентилиденовый лиганд из двух неопентильных лигандов в результате реакции отщепления -водородного атома в гипотетическом Mo(NR’)(CH2-t-Bu)2(Triflate)2 интермедиате 49. Имидные (NR’) и алкоксидные группы можно варьировать в широких пределах, и поэтому становится возможным синтез большого числа катализаторов на основе молибдена. Молибденовые бисалкоксидные катализаторы, к тому же являются заметно более активными в большом числе реакций метатезиса олефинов, в особенности, если алкоксидом является сильная электронооттягивающая группа OCMe(CF3)2, а молибденциклобутановый интермедиат становится менее стабильным, чем вольфрамциклобутановый интермедиат в отношении отщепления олефина. Стерически объемистые OCMe(CF3)2 и Ar (2,6-диизопропилфенил) группы играют также существенную роль в предотвращении бимолекулярных реакций между промежуточно образующимися частицами Mo(NAr)(CHR’)(OR)2 во многих таких реакциях. Постепенно мы начали понимать ту меру, до которой мы можем управлять реакцией метатезиса олефинов путем систематического варьирования размера и электронных характеристик алкоксидных и имидных групп. Мы обнаружили также, что могут существовать два изомера любого катализатора M=CHR (схема 20). В том изомере, образование которого обычно наблюдается, алкилиденовый заместитель направлен в сторону имидной группы (син-), тогда как в другом изомере он направлен в сторону от имидной группы (анти-). |
|
 |
|
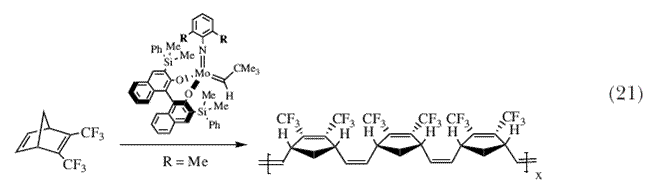
Син- и анти-изомеры могут образовываться в результате протекания реакции метатезиса, однако син- и анти-изомеры любого данного алкилидена также легко могут взаимно превращаться друг в друга в результате вращения вокруг связи Mo=C. Было найдено, что скорость вращения неопентилидена или неофилидена изменяется как минимум в 106 раз, и зависит (в значительной степени) от природы алкоксида 50,51, причем скорость вращения относительно велика в присутствии алкоксидных лигандов, таких как трет-бутоксильные, и относительно невелика в присутствии гексафтор-трет-бутоксильных лигандов. При изучении реакций полимеризации с раскрытием цикла в результате метатезиса (ROMP), одной из множества разновидностей основной реакции метатезиса олефинов, мы обнаружили (для одного типа полимеризуемых мономеров), что анти-изомер был более реакционноспособен примерно в 104 раза. Поэтому в каталитической реакции могут участвовать тот или другой, или оба – син- и анти-интермедиаты, с результатом, зависящим от природы имидной и алкоксидной групп, а также от реакционной способности олефина, используемого в реакции с син- и анти-интермедиатами, а также от температуры и других переменных. Эти обстоятельства в определенной степени осложняют детальное понимание катализа реакции метатезиса такими катализаторами, однако в то же самое время существенным образом увеличивают гибкость катализаторов и вероятность того, что реакция метатезиса пройдет успешно. Реакции полимеризации циклических олефинов с раскрытием цикла в результате метатезиса начал изучаться за много лет до создания катализаторов с четко установленным строением 17,18. Мы естественно задавали себе вопрос о том, не могут ли катализаторы с четко установленным строением могут вести себя как катализаторы полимеризации циклических олефинов, и если это так, какими преимуществами они обладают? В нашей недавней совместной публикации, явившейся итогом моего творческого отпуска в Калтехе в качестве стипендиата Fairchild в 1986, Грубс и я показали, что эти новые катализаторы с известной структурой ведут себя предсказуемым образом, и что процесс в соответствующих условиях может быть «живым», т.е., промежуточные алкилидены, содержащие растущую полимерную цепь, не разлагаются 52. В моей лаборатории мы намеревались показать, что процесс полимеризации можно контролировать существенным образом, получая полимеры с высоко регулярной повторяющейся структурой. Как правило, полимеры с высоко регулярной структурой имеют более интересные свойства и поэтому их получение более предпочтительно. Во многих случаях мы обнаружили, что всю структуру полимера можно проконтролировать до мелочей, однако самым важным являются изменения в структуре катализатора. Например, (схема 21), если катализатор содержит специальный бинафтолятный лиганд и диметилфенилимидную группу (R = CH3 в 2,6-дизамещенной фенильной группе), то образуется высоко регулярный цис-, изотактичный полимер 53,54. |
|
 |
|
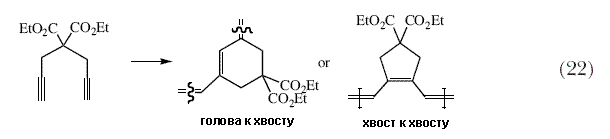
| Напротив, если группа R в имидном лиганде является изопропильной, тогда полимер имеет относительно случайную структуру. Считается, что меньшая по объему диметилфенилимидная группа позволяет протекать реакции полимеризации исключительно через син-изомеры с образованием цис-связей. Возможность управления «изотактичностью», или тем, в какую сторону повернуты пятичленные циклы в этой полимерной цепи, является наибольшей в присутствии хирального (рацемического) бинафтолятного лиганда, что является следствием «эффекта энантиоморфного сайта» в ходе процесса полимеризации. В серии экспериментов с родственными мономерами, содержавшими две сложноэфирные группы, полученными с использованием энантиомерно чистых спиртов, мы получили возможность различать изотактические и синдиотактические полимеры, и следовательно могли выявлять цис-, изотактические структуры, которые образовывались в результате эффекта энантиоморфного сайта, и транс-синдиотактические структуры, которые образовывались в результате эффекта конца цепи, в первую очередь в ROMP-полимерах 55. Степень, в которой можно управлять строением полимера в результате систематической, тонкой настройки структуры катализатора имеет большое значение для синтеза большого числа ROMP-полимеров, которые в настоящее время получают с использованием в качестве инициаторов алкилиденовых комплексов металлов в высокой степени окисления. Некоторе время тому назад стало известно, что «классические» Mo и W катализаторы, которые катализируют реакцию метатезиса олефинов, катализируют также реакции полимеризации алкинов. Было высказано предположение, что за это ответственны алкилидены металлов 56-58. Мы показали, что комплексы с известной структурой действительно вызывают полимеризацию алкинов 59 или циклополимеризацию 1,6-гептадиинов 60 (схема 22) с образованием полимеров, аналогичных тем, что описаны в литературе. | |
 |
|
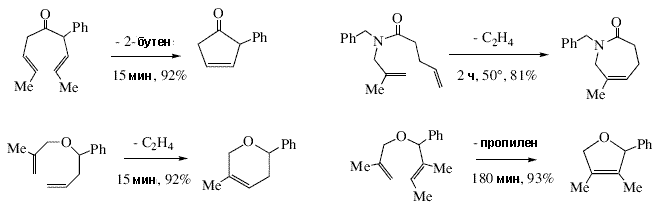
| В случае реакции циклополимеризации 1,6-гептпдиинов, таких как диэтилдипропаргилмалонатов, могут образоваться шестичленные или пятичленные циклы, в зависимости от региохимии реакции присоединения первой молекулы алкина к алкилиденовому комплексу. Поскольку оптические свойства таких полиенов меняются в зависимости от строения полимера и длины цепи 61, весьма желательно получить полимер с однозначной структурой и регулировать молекулярный вес такого полимера путем проведения реакции полимеризации в присутствии катализатора с точно установленным строением. Хотя работы в этой области только начинаются, можно утверждать, что это вполне возможно 62,63. В 1992 Г. К. Фу и Р. Х. Грубс опубликовали две статьи, в которых они показали, как Mo(NAr)(CHCMe2Ph)[OCMe(CF3)2]2 можно использовать для быстрого и эффективного получения циклических олефинов, содержащих функциональные группы, отличные от двойных C=C связей 64,65, с образованием только одного побочного продукта, являющегося летучим олефином, таким как этилен, пропилен или бутен (схема 5). Можно получать циклы различного размера, и даже циклы, содержащие тетразамещенные олефиновые связи. Эти статьи помогли обратить внимание химиков-органиков к тому, что возможности, которыми обладает реакция метатезиса олефинов с участием катлизаторов с известной структурой, имеют большой потенциал в органической химии. Тот факт, что Mo и W катализаторы этого общего класса, чувствительны к воздуху, влаге, и в определенной степени к начилию функциональных групп, не мешают их использованию в органических рекциях. | |
 |
|
| Рисунок 5. Некоторые реакции метатезиса с замыканием цикла, инициируемые Mo(NAr)(CHCMe2Ph)[OCMe(CF3)2]2. | |
| В 1995 A. Х. Ховейда с группой своих сотрудников сообщили о синтезе циклического природного соединения, флувирицина-B1, в котором одна из стадий заключается в замыкании цикла в результате реакции метатезиса (RCM) (схема 23) 66. В этой статье показано, что довольно сложные молекулы могут быть получены с использованием реакции метатезиса. | |
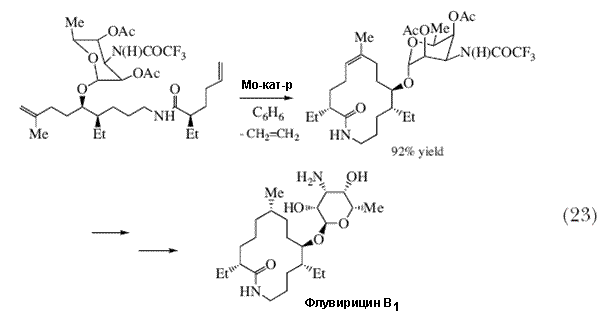 |
|
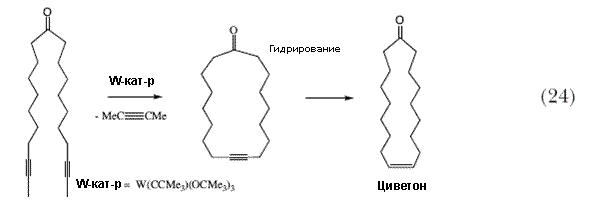
Это и другие применения реакции метатезиса, наряду с коммерческой доступностью Mo(NAr)(CHCMe2Ph)[OCMe(CF3)2]2, помогли развить применения реакций метатезиса, катализируемых комплексами Mo в органической химии 67. Я полагаю, что уверенность в том, что метатезис может быть использован в органической химии в качестве рядовой химической процедуры, явилось значительным фактором, который привел Грубса к разработке рутениевого катализатора в начале 1990 годов 20,68,69. Использование катализаторов метатезиса алкинов в органической химии также было реализовано в 1990 годах, вначале благодаря работам А. Фюрстнера, который показал, что с помощью метатезиса алкинов с (Me3CO)3W Наблюдается появление большого числа других реакций, в которых образование тройной углерод-углеродной связи по реакции метатезиса алкинов используется как часть синтетической процедуры, например, синтез цитотоксичного алкалоида морского происхождения мотупорамина C 72, простагландина E2-1,15-лактона 73, и эпотилона A и C, каждый из которых содержит в цикле цис-двойную олефиновую связь (рисунок 6)74. |
|
 |
|
| Рисунок 6. Некоторые молекулы, синтез которых включает в себя метатезис алкинов для образования цис-двойную связь. | |
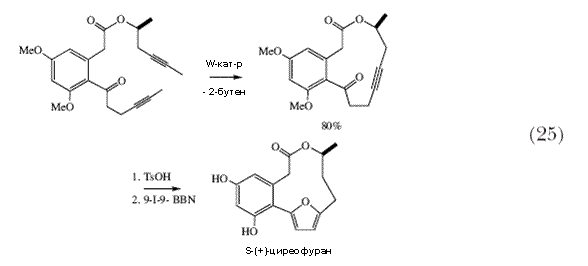
| Метатезис алкинов может быть полезным и в других случаях, как это показано в синтезе S-(+)-циреофурана (схема 25) 75. Хотя катализаторы метатезиса алкинов на базе молибдена были известны ранее 76,77, работами Куммниса 78, Фюрстнера 79 и Мура 80 недавно было показано, как эти сравнительно труднодоступные Mo катализаторы могут быть получены in situ из трисамидоалкилидиновых предшественников. | |
 |
|
| На основании знаний того, что определенные стерически защищенные бифеноляты и бинафтоляты можно присоединить к Mo с образованием стабильных неопентилиденовых или неофилиденовых инициаторов ROMP, в середине 1990 годов мы обратились к разработке энантиомерно чистых катализаторов реакций асимметрического метатезиса. В 1997 г. были получены некоторые предварительные результаты с энантиомерно чистым бифенолятным катализатором (рисунок 7). Затем мы начали сотрудничать с A. Х. Ховейда, что имело своей целью использование реакций асимметрического метатезиса в органическом синтезе 67,81,82. | |
 |
|
| Рисунок 7. Строение Mo(NAr)(CHCMe2Ph) (3,3’-ди-трет-бутил-5,5’,6,6’-тетраметил-1,1’-бифенил-2,2’-диолята). | |
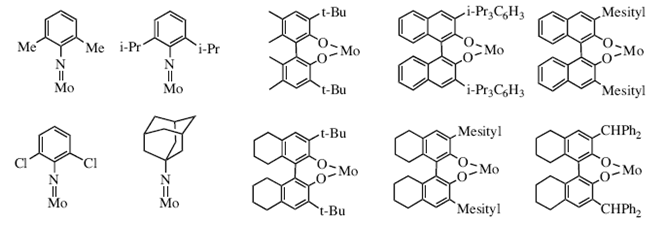 |
|
| Рисунок 8. Имидо-группы и энантиомерно чистые диоляты (показаны в рацемической форме), применяемые для получения асимметрических катализаторов. | |
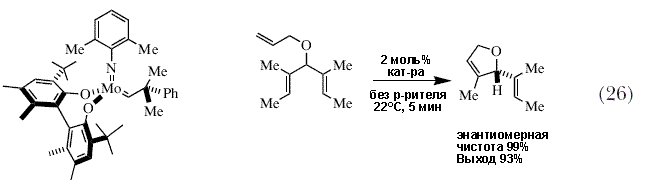
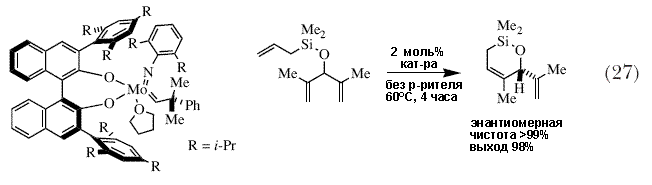
| Модульный принцип сборки катализаторов позволил нам получить большое число энентиомерно чистых вариантов, содержащих один из некоторых имидных лигандов и различные диоляты (рисунок 8). Скоро стало очевидным, что при выборе правильного катализатора, асимметричные реакции могут быть эффективны с точки зрения как выхода, так и энантиомерной чистоты, во многих случаях с довольно быстрым образованием единственного энантиомерно чистого продукта с практически количественным выходом (схемы 26 и 27) 83. Опять-таки, в качестве побочных продуктов обычно получаются простые олефины, такие как этилен, пропилен или бутен. | |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
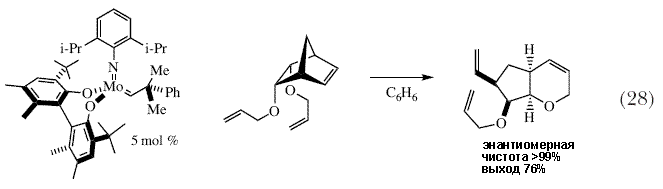
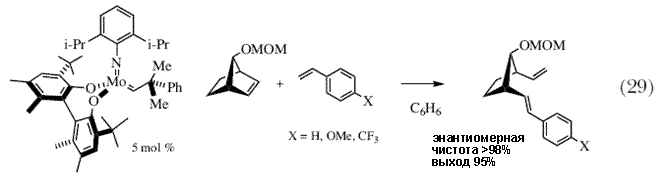
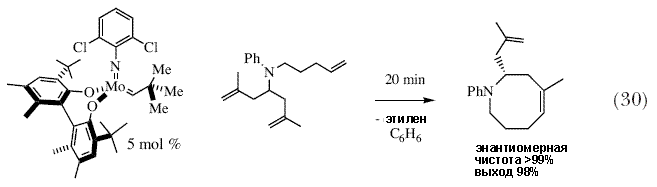
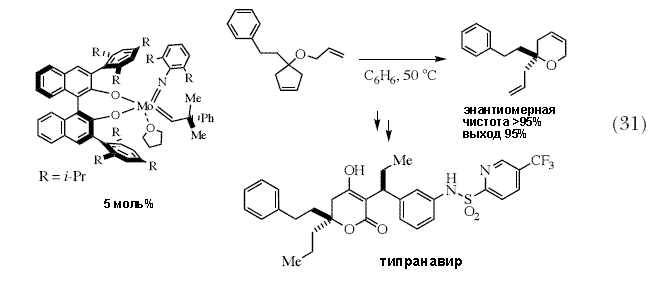
| Был разработан широкий набор других ассиметрических синтезов, среди них метатезис с раскрытием цикла/замыканием цикла (схема 28) 84, перекрестный метатезис с раскрытием цикла (схема 29) 85, и замыкание цикла третичных аминов с образованием продуктов, напоминающих подструктуры различных лекарств и других природных соединений (схема 30) 86. Было также показано, что синтез лекарственных препаратов, таких как трипанавир, ингибитор протеазы HIV, можно существенным образом сократить в результате использования стадии асимметрического метатезиса для получения проблематичного энантиомерно чистого третичного эфира (схема 31) 87,88. Однако это всего лишь несколько примеров того, что было сделано за последние несколько лет. | |
 |
|
 |
|
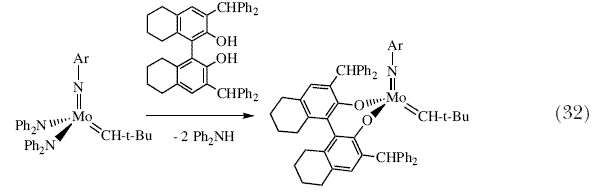
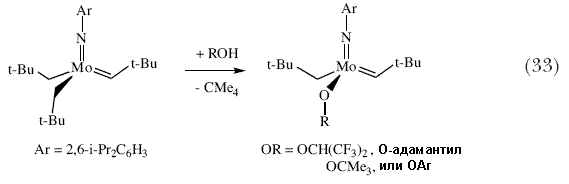
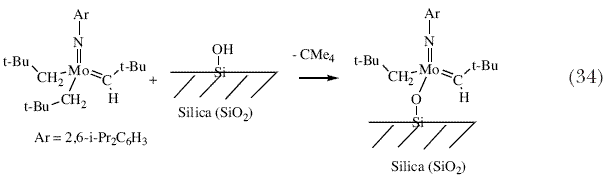
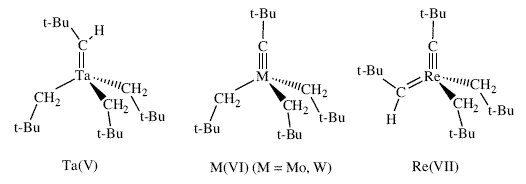
| Выделение и испытания большинства модификаций катализатора для какой-либо специфической асимметрической реакции показывает, что эффективность асимметрического катализатора с точки зрения выхода и энантиоселективности может существенным образом меняться, и при условии достаточно продолжительного поиска можно быть более или менее уверенным в нахождении эффективного катализатора. Однако, необходимо получить, выделить и испытать множество катализаторов. Поэтому, мы начали исследовать возможность получения катализаторов in situ, о которых нам известно, что они существуют в виде относительно устойчивых комплексов, либо путем добавления бифенолятов или бинафтолятов к бистрифлатным предшественникам (схема 19), или путем добавления исходных бифенолов или бинафтолов к бисамидным предшественникам (схема 32). Мы показали, что катализаторы, полученные in situ для асимметрического метатезиса ведут себя точно также, как и катализаторы , которые были выделены и очищены 89. Неожиданно, аналогичные подходы с использованием динеопентильных молекул привели к образованию моноалкоксидных катализаторов (схема 33) 90. Вначале мы подумали, что они будут сравнительно слабыми катализаторами метатезиса, поскольку динеопентильные молекулы значительно менее активны в реакции метатезиса. Поэтому присутствие одного неопентила и одного алкоксида вместо двух алкоксидов должно быть пагубным. Однако первоначальные результаты показали, что моноалкоксиды являются высокоактивными катализаторами. Недавние теоретические расчеты в подобных изоэлектронных рениевых системах позволили предположить, почему это может иметь место 91. Образование моноалкоксидов из динеопентильных копмплексов недавно сделало возможным осуществление синтеза чрезвычайно активного молибденового (а также вольфрамового) катализатора реакции метатезиса с «известным строением» на поверхности силикагеля (схема 34), который оказался сравнительно долгоживущим, поскольку промежуточные алкилидены не могут разлагаться бимолекулярно 92. В настоящее время можно получить множество катализаторов с более или менее известным строением на поверхности силикагеля с использованием других Ta(V), Mo(VI), W(VI) и Re(VII) неопентильных прекурсоров (рисунок 9) 93. В некоторых случаях было показано, что можно наблюдать протекание совершенно новых реакций, например «метатезис алканов» при использовании танталовых катализаторов 94. Эта реакция, как было показано, включает в себя стадии метатезиса алкенов 95. Рений образует катализаторы метатезиса «классического» типа. Поэтому мы почувствовали, что нам удастся получить такие катализаторы с известным строением. | |
 |
|
| Рисунок 9. «Чистые» источники катализаторов, образующихся после прибавления их на поверхность силикагеля. | |
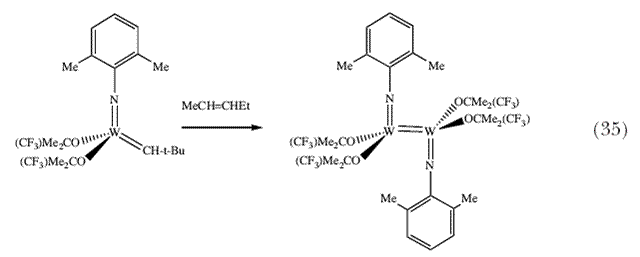
| С целью сохранения четырехкоординированной геометрии вокруг нейтрального Re центра, необходим стерически объемистый лиганд, который был бы связан тройной связью с металлом. Единственным логическим выбором является неопентилидиновый лиганд. Действительно, хорошо охарактеризованный бисалкоксидные катализаторы на основе Re(VII) для метатезиса алкенов можно получить, следуя принципам, почерпнутым из химии W и Mo 96,97, поскольку неопентилидиновый лиганд лишь с трудом реагирует с олефинами (схема 10). Считается, что моноалкоксидные неопентильные аналоги являются даже более реакционноспособными, чем бисалкоксиды, в особенности на поверхности силикагеля 91. И наконец, мы обнаружили, что могут образовываться двойные связи M=M, где M = Mo 98, W 99 или Re 100 как следствие разложения алкилиденовых комплексов (схема 35). Двойные связи металл-металл являются необычными, в особенности если эти двойные связи не сопровождаются образованием мостиковых связей с участие потенциально способных к образованию таких связей лигандов, например, алкоксидных или имидных групп. | |
 |
|
Тот факт, что эти комплексы сами по себе катализируют метатезис некоторых олефинов 98 (медленно) указывает на возможность того, что алкилидены получаются из соединений с M=M связями. Если окажется, что это так, это будет строгим доказательством того, что взаимопревращение связей металл-металл, углерод-углерод и металл-углерод возможно в случае двойных связей, так как это нам известно в случае тройных связей (схема 17). За последние 30 лет мы прошли огромный путь, от катализаторов реакции метатезиса «неизвестного строения» к тем, чьей структурой и реакционной способностью в растворе (а теперь это возможно и на поверхности) мы можем управлять с большой точностью. Остаются фундаментальные проблемы с известными катализаторами, наиболее важной является то, как предотвратить разложение катализатора и/или как регенерировать катализаторы из «чистых» продуктов разложения. Я ожидаю, что эти вопросы найдут свое решение и надеюсь, что результаты синтеза и фундаментальных исследований новых типов катализаторов, а также их применение к решению широкого круга проблем, будет продолжать оставаться ощутимым в будущие годы. |
|
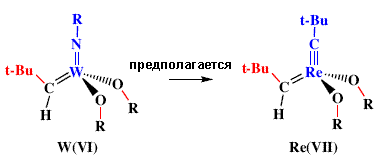 |
|
| Рисунок 10. Неопентилидиновые бисалкоксидные комплексы рения являются катализаторами метатезиса олефинов. | |
| БЛАГОДАРНОСТИ | |
| Я благодарен за свободу, данную мне в начале моей работы в Центральном Исследовательском Департаменте комппании duPont, за возможность расширить мои усилия и мои интересы в MIT, и за поддержку, предоставленную различными фондами (National Science Foundation, National Institutes of Health, Navy, Army и Департаменту энергетики) в течение всех лет занятий химией металлорганических соединений с металлами в высокой степени окисления, а также другими проектами. Я нахожусь в вечном долгу перед моими дипломниками и аспирантами, каждый из которых соим собственным способом способствовал тем огромным усилиям, потребовавшимся для перехода от нескольких наблюдений и новых соединений к химии, которая дала значительный импульс исследованиям в областях вне неорганической химии. И наконец, я хочу поблагодарить мою жену Нэнси и мою семью за их любовь и поддержку. | |
| ЛИТЕРАТУРА | |
1. G. Wilkinson, Нобелевская лекция, декабрь 11, 1973. |
|
| Портретный снимок Ричарда Р. Шрока – фотограф – Б. Хезерингтон. | |