ЛОГИКА ХИМИЧЕСКОГО СИНТЕЗА:
МНОГОСТАДИЙНЫЙ СИНТЕЗ СЛОЖНЫХ КАРБОГЕННЫХ МОЛЕКУЛ
Нобелевская лекция, 8 декабря, 1990
ЭЛИАС ДЖЕЙМС КОРИ
Химический факультет, Гарвардский Университет, Кембридж, Массачусетс, США
Карбогены, члены семейства углерод содержащих соединений, могут существовать в виде бесконечного многообразия структур, форм и размеров. Встречающиеся в природе карбогены, или органические вещества, как традиционно их называют, составляют основу всей жизни на Земле, и наука о б этих веществах на молекулярном уровне определяет фундаментальный язык этой жизни. Химический синтез этих встречающихся в природе карбогенов и многих миллионов неприродных карбогенных веществ было одним из основных рискованных предприятий науки в этом столетии. Этот факт подтверждается присуждением Нобелевской премии в химии в 1990 г. за «создание теории и методологии органического синтеза». Химический синтез занимает уникальное положение в сердце химии, центральной науки, и его влияние на нашу жизнь и общество является всепроникающим. Например, многие современные медицинские препараты являются синтетическими, и многие препараты будущего разрабатываются, и будут производиться химиками-синтетиками. К области синтетической химии принадлежит массив ответственности, который является критичным для будущего человечества, не только в отношении здоровья, материальных и экономических потребностей нашего общества, но также и для достижения понимания строения материи, химических изменений и жизни на самом высоком уровне, на который только способен человеческий разум.
Период после Второй мировой войны охватывает значительные достижения в химическом синтезе. В первые два десятилетия этого периода были разработаны химические синтезы, которых было трудно ожидать в начале этого столетия. Во-первых, в результате тщательно продуманных многостадийных процессов были собраны очень сложные молекулы, например, витамин A (О. Айслер, 1949), кортизон (Р. Б. Вудворд, Р. Робинсон, 1951), морфин (М. Гейтс, 1956), пенициллин (Д. К. Шихан, 1957), и хлорофилл (Р. Б. Вудворд, 1960).1 Этот поразительный скачок вперед, который был отмечен присуждением Нобелевской премии по химии Р.Б. Вудворду в 1965 2, сопровождался столь же значительными научными успехами в последние три десятилетия, в ходе которых химический синтез поднялся на качественно более высокий уровень сложности. В настоящее время, во многих лабораториях по всему миру химики с изумительной скоростью синтезируют сложные карбогенные структуры, которые практически невозможно было получить в 1950-е годы или в начале 1960-х. Этот прогресс был инициирован благодаря доступности более мощных концептуальных процессов планирования химического синтеза, использованию новых химических методов, в виде реакций и реагентов, и появлению усовершенствованных методов анализа, разделения и определения строения. Многие талантливые исследователи по всему миру внесли свой вклад в последние достижения химического синтеза. Их усилия составили коллективный проект огромного размера, даже если и выполнялись независимо, и их идеи и открытия вступали в синергетическое взаимодействие ко всеобщей пользе. Я счастлив тому, что избран Нобелевским комитетом за вклад в науку химического синтеза, однако я в еще большей степени рад тому, что эта важная область науки вновь получила столь высокое признание.
Генезис
Весной 1947, будучи студентом Массачусетского Института, я проходил курс синтетической органической химии повышенного уровня, преподававшийся известным химиком А.К. Коупом, в котором рассматривались основные синтетические реакции. Нам объяснили, что осталось найти лишь небольшое число методов синтеза, поскольку за предшествующие пятьдесят лет было открыто только пять важных реакций; и нам, студентам, было предписано изучить, как разрабатывать химические синтезы, используя доступных набор известных построений. Нам давали множество молекулярных структур в качестве синтетических задач. После выполнения нескольких наборов задач, я приобрел достаточный навык и опытность, для того чтобы с легкостью справиться с оставшимися заданиями, почти так же, как это было с изучением английского языка, доказательством математических теорем или с игрой в шахматы. Мои вновь обретенные умения в решении химических задач по всей видимости являлись скорее результатом знания некого автоматического «ноу-хау» нежели итогом точного применения определенных методик. Однако, несмотря на то, что я овладел классическими реакциями, разработка синтезов молекул, выходящих за рамки умеренного уровня сложности этих учебных задач, мне не удавалась. Такие молекулы как морфин, холестерол, пенициллин или сахароза были настолько неприступными, что они определяли границы возможного в 1947; каждая из них казалась уникальной и требовала очень высокого уровня креативности и изобретательности. В течение многих лет основная часть моих исследований была направлена на зондирование этих границ и повышение уровня синтетической науки и этот подход состоял из трех основных компонентов: разработка более общих и эффективных способов мышления в области синтетических задач, разработка новых общих реакций и реагентов для синтеза, а также разработка и проведение эффективных многостадийных синтезов сложных молекул на пределе возможностей современной синтетической науки.
Ретросинтетический анализ
В течение первой половины этого столетия большинство синтезов были разработаны путем выбора подходящих исходных веществ, после поиска методом проб и ошибок из числа коммерчески доступных соединений, имеющих структурное сходство с целью синтеза. Затем рассматривались подходящие реакции для превращения выбранных исходных веществ в желаемый продукт. В большинстве случаев синтетическое планирование сильно зависело от принятой точки старта. Весной 1957 мне в голову пришла простая идея, которая привела к полностью отличному способу разработки схем химического синтеза. В этом подходе целевая структура подвергается процессу деконструкции, который соответствует обратной реакции синтеза, с целью превращения этой целевой структуры в более простые молекулы прекурсоров, без каких либо предположений относительно исходных веществ. Каждый из полученных таким образом прекурсоров затем исследуется тем же способом, и процесс повторяется до тех пока в итоге не получатся простые или коммерчески доступные продукты. Эта «ретросинтетическая» или «антитетическая» процедура составляет основу общей логики синтетического планирования, которая была разработана и продемонстрирована в действии в течение последующего десятилетия.3,4,5 В одном из первых примеров ретросинтетического планирования трициклического сесквитерпена лонгифолена (1) (Рис. I) были разработаны несколько привлекательных путей синтеза, один из которых был выбран и подтвержден в результате экспериментального выполнения. 6 Основные идеи ретросинтетического анализа были использованы для разработки большого числа других синтезов, а также для создания компьютерной программы для генерирования возможных путей синтеза сложных целевых структур без задания каких либо потенциальных исходных веществ или промежуточных веществ для синтеза.4,5 Принципы ретросинтетического анализа недавно были обобщены в учебнике «Логика химического синтеза»7, который был написан для студентов-дипломников и аспирантов химиков. Ретросинтетический способ осмысления химического синтеза предоставляет также логичный и эффективный путь обучения планированию синтеза начинающих студентов и студентов-дипломников, хороший пример тесной связи между обучением и исследованиями в академическом институте. А теперь будет приведен подробный обзор ретросинтетического планирования синтеза.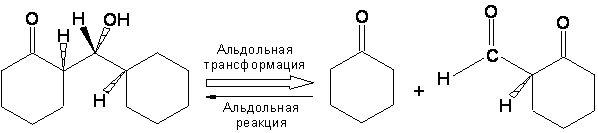
Трансформации отличаются друг от друга своей способностью упрощать целевую молекулу. Наиболее мощные из упрощающих трансформаций, которые снижают сложность молекулы в ретросинтетическом направлении, занимают особое положение в иерархии всех трансформаций. Их применение, даже если соответствующий ретрон отсутствует, может исключить использование большого числа не упрощающих трансформаций для получения такого ретрона. В общем, назначение упрощающих трансформаций заключается в модификации структурных элементов, которые ответственны за сложность молекулы: размер молекулы, сочленение циклов (топология), наличие стереоцентров, наличие элементов и функциональных групп, химическую реакционную способность, структурную нестабильность, и плотность размещения усложняющих элементов. Сложность молекулы важна для выбора стратегии. Для каждого типа сложности молекулы имеется набор общих стратегий для преодоления этой сложности. Например, в случае сложной полициклической структуры, стратегии упрощения скелета молекулы, т.е. топологические стратегии, должны играть важную роль в выборе трансформации. Однако наиболее эффективный способ ретросинтетического анализа заключается не в отдельном использовании отдельных стратегий, а в конкурентном применении столь большого числа различных независимых стратегий, насколько это возможно. Основные типы стратегий7, которые имеют значение в ретросинтетическом анализе, могут быть сформулированы в самом общем виде следующим образом.8
1. Стратегии, основанные на трансформациях - стратегический поиск или прогнозирование с целью применения мощной упрощающей трансформации (или тактические комбинации упрощающих трансформаций) к TGT с определенными требуемыми ключевыми свойствами. Ретрон, необходимый для применения эффективной трансформации, может отсутствовать в сложной TGT, и для его установления может понадобиться большое число антитетических стадий (подцелей).
2. Стратегии, нацеленные на структуру - направлены на структуру потенциального интермедиата или потенциального исходного соединения. Такая цель существенно ограничивает ретросинтетический поиск и делает возможным применение двунаправленной технологии поиска.
3. Топологические стратегии - идентификация одного или нескольких индивидуальных расчленений связей или взаимосвязанных пар связей в качестве стратегических. Топологические стратегии могут также привести к идентификации ключевых субструктур для разборки молекулы или к использованию трансформаций, обратных перегруппировкам.
4. Стереохимические стратегии - общие стратегии, которые удаляют стереоцентры и стереовзаимосвязи под стереоконтролем. Такой стереоконтроль может возникать из контроля механизма трансформации или субстратно-структурного контроля. В случае первого ретрон для отдельной трансформации содержит существенную стереохимическую информацию (абсолютную или относительную) по одному или большему числу стереоцентров. Стереохимические стратегии могут также диктовать сохранение определенных стереоцентров в ходе выполнения ретросинтетической процедуры или соединение атомов в трехмерной близости. Основная функция стереохимических стратегий является достижение экспериментально подтверждаемого исчезновения стереоцентров, включая исчезновение молекулярной хиральности.
5. Стратегии основанные на функциональных группах. Ретросинтетическое понижение сложности молекулы, содержащей функциональные группы (FG), принимает различные формы. Отдельные FG или пары FG (а также соединяющая их цепь атомов) могут непосредственно указать на место расчленение скелета TGT с образованием более простых молекул или сигнализировать о применении трансформаций, которые замещают функционльные группы на атом водорода. Замена функциональных групп (FGI) является обычно применяемой тактикой для генерирования ретронов упрощающих трансформаций из TGT. FG могут указывать на трансформации, которые стереоселективно удаляют стереоцентры, топологически разрывают стратегические связи или связывают близко расположенные атомы с образованием циклов.
6. «Другие» типы стратегий. Распознавание субструктурных фрагментов в TGT, которые являются главными препятствиями для синтеза, часто дает основной стратегический посыл. Некоторые другие стратегии возникают из требований частной задачи, например, требование того, чтобы несколько родственных целевых структур можно было синтезировать из одного общего интермедиата. Молекула TGT, которая противостоит ретросинтетическому упрощению, может потребовать изобретения новой химической методологии. Распознание препятствий для синтеза дает стимул для открытия таких новых процессов. Важным является применение цепи гипотез для того чтобы направить поиск эффективного направления ретросинтетического анализа. Другие стратегии направлены на оптимизацию дизайна синтеза после того, как ретросинтетически уже был сгенерирован набор путей, специально для упорядочения стадий синтеза, использования стадий защиты или активации, или определения альтернативных путей.
Систематический и скрупулезный ретросинтетический анализ является общим принципом решения синтетических задач, среди которых индивидуальные стратегии занимают важное место. Другой общей идеей является одновременное использование такого количества независимых стратегий, насколько это возможно, для направления поиска ретросинтетических путей. Чем больше число стратегий, которые используются параллельно для создания направления анализа, тем, вероятно, будет проще анализ и тем проще будет искомый план синтеза.10
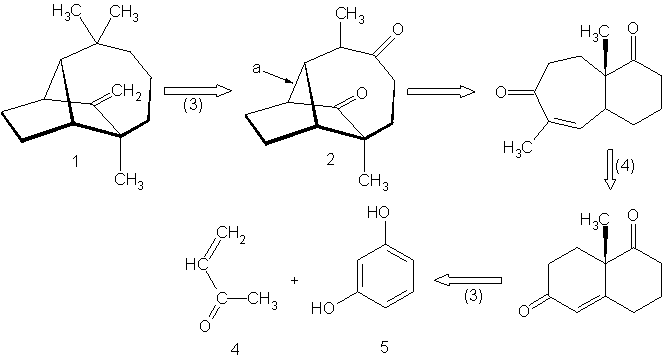
Ретросинтетический анализ лонгиофолена
Рисунок I
Компьютерный ретроспективный анализ
Использование компьютеров для генерирования возможных путей химического синтеза, которое было впервые продемонстрировано в 1960-е годы4,5,9,11, стало возможным благодаря созданию ретросинтетических методов, описанных выше и необходимой компьютерной методологии. Графический ввод структур рисованием от руки с использованием электростатических планшетов и стилуса, привычным для химика образом, и вывод разультата на видеотерминал,12 дает чрезвычайно простой и эффективный интерфейс между химиком и машиной. Химические структуры в машине были представлены в виде посредством таблиц атомов и связей, и обрабатывались посредством соответствующих инструкций. Были изобретены алгоритмы для восприятия машиной структурных свойств, модельных структур и фрагментов, которые необходимы для синтетического анализа. Были созданы технологии сохранения и извлечения информации о химических трансформациях (включая распознавание и манипуляция с ретронами) с использованием «химического английского языка» высшего уровня. Была разработана интерактивная программа (LHASA), с любым уровнем управления или ввода, необходимого пользователю, и способная к эмулированию подходов к решению задачи химика-синтетика. Химическая база данных, написанная так, чтобы быть понятной химику-практику, содержит все типы информации, необходимой для генерировании и оценки ретросинтетических изменений, например, данные по индивидуальным трансформациям и их механизмам, границах применимости и ограничениях. Эффективность программы ниже той, которая в конечном итоге была бы возможной, вследствие необъятности задачи и ограниченности объема наших исследовательских усилий. Однако, несмотря на существующие ограничения, включая скромный размер базы данных по примерно 2000 трансформациям, LHASA способна выдать интересные предположения о путях синтеза нужных целевых молекул. Настоящий уровень возможностей LHASA лучше всего можно оценить по результатам ее работы над какой либо специальной задачей. На рис. II показано EXTGT-дерево, сгенерированное программой LHASA для антивирусного препарата афидиколина, при использовании только одного частного направления анализа. В этом древе путь от интермедиатов 332 и 333 к афидиколину состоит из структур, показанных на рис. III.13 Предложение программы LHASA таких неочевидных путей является одновременно как стимулирующим, таки и значимым для химика. Область компьютерного синтетического анализа зачаровывает сама по себе, и конечно же является одной из наиболее интересных задач в области компьютерного интеллекта.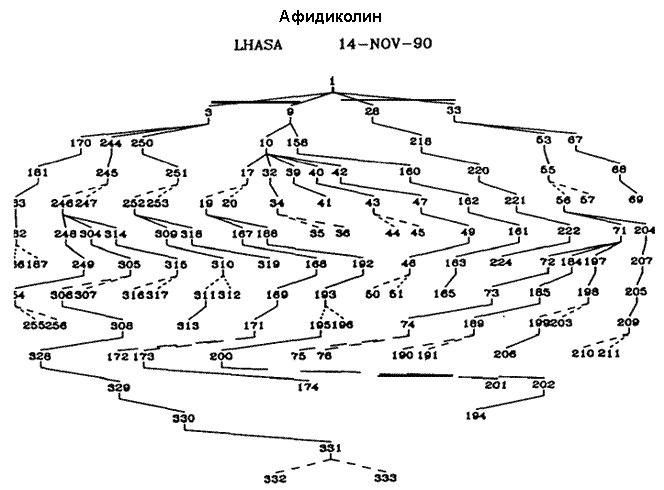
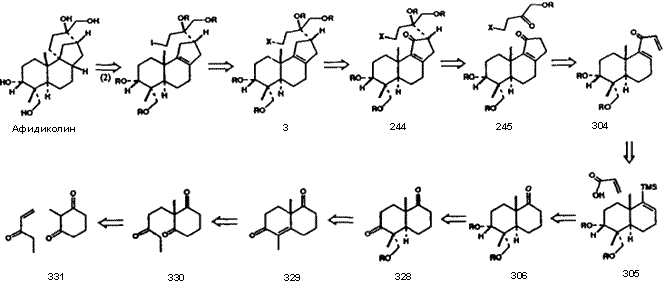
Рисунок III
Вследствие огромного объема памяти и высокого быстродействия современных компьютеров и высокая вероятность продолжения развития в этом направлении, становится ясным, что компьютеры могут играть важную роль в синтетическом дизайне. Однако прежде чем этот потенциал может быть реализован, должны быть решены многие сложные вычислительные проблемы. Многостадийный ретросинтетический прогноз, даже под стратегическим управлением, требует наличия сложного и мощного программного обеспечения. Огромное количество информации - стереохимической и химической - должно быть сгенерировано и проанализировано с использованием всего доступного химического знания. Требуется создание крупного проекта.
Новая синтетическая методология и многостадийный синтез сложных молекул
Открытие новых реакций и реагентов революционировало область карбогенного синтеза, буквально разместив чрезвычайно мощную новую химию рядом с классическими реакциями периода до 1950 г. Без этой методологии осуществление современных химических синтезов было бы невозможным. Две ранних вехи в этом развитии: открытие Виттигом синтеза олефинов и реакции гидроборирования олефинов, были отмечены присуждением Нобелевской премии по химии за 1979 Г. Виттигу и Х.К. Брауну. Последние разработки дали большое число методов, которые примечательны своей высокой химической селективностью и стереохимическим контролем, а также своей пригодностью для конструирования сложных молекул. Фактически многие новые синтетические реакции были открыты в результате насущной необходимости в связи со специфическими задачами, включая синтез новых или замысловатых структур и целенаправленный поиск подходящей методологии. Рациональный дизайн таких методов зависит от использования теории механизмов химических реакций, принципов стереохимии и широкой области химии, включающей в себя большое число элементов и короткоживущих интермедиатов. Ключом к успеху большого числа многостадийных химических синтезов, которые были проведены в нашей лаборатории в течение ряда лет, было изобретение новой методологии. Впоследствии, более пятидесяти таких методов были разработаны в нашей лаборатории, этот аспект синтетических исследований невозможно охватить в кратком сообщении. Однако несколько примеров могут послужить иллюстрацией эффективности этих новых методов, открывающих доступ к получению редких и ценных карбогенным соединениям, а также их влияние на всю область химического синтеза.Открытие и идентификация в 1967 г. ювенильного гормона насекомых Cecropia, в настоящее время известного как JH-I)14 вызвало огромный интерес вследствие возможности использования таких нетоксичных соединений для управления численностью популяций насекомых. Химический синтез был необходим вследствие крайней ограниченности исходного материала из природных источников. Несмотря на кажущуюся простоту структуры 6, стереоспецифичный путь синтеза был неочевиден, поскольку в 1967 г. не существовало общих методов для стереоконтролируемого получения трехзамещенных олефиновых фрагментов, которые содержит данная молекула. Первый стереоспецифичный синтез 616 стал возможен благодаря использованию новой методологии, которая была специально разработана для решения этой задачи. Сокращенная версия синтеза показана на рис. IV. Первый олефиновый интермедиат (7) был синтезирован из пара-метокситолуола способом, который гарантировал Z-конфигурацию стереоцентров. Реакция 8 с LiAlH4 (транс-гидроалюминирование) с последующей обработкой иодом (замещение Al на I) стереоспецифично дает 9 в результате новой последовательности превращений16. Замещение иода в 9 на этильную группу было осуществлено с помощью другой новой реакции, кросс-сочетания винилиодида с органилмедным реагентом, в результате чего стереспецифично был получен 10. Полностью аналогичная серия реакций позволила превратить 10 в триен 11, из которого JH-I (6) был получен с помощью новой последовательности реакций селективного окисления. Таким образом, синтез, приведенный на рис. IV, зависел не менее чем от четырех новых синтетических методов. Три из этих методов вошли в самую широкую практику. Сочетание углеродных фрагментов с использованием органилмедных реагентов в настоящее время является основным методом химического синтеза17. Родственная реакция карбометаллирования ацетиленов, также разработанная в связи с синтезом 6,16b,18 получила развитие во многих направлениях, и этот подход стал самым обычным для стереоспецифичного построения трехзамещенных двойных связей, часто встречающегося типа структурных единиц в биологически активных природных соединениях.
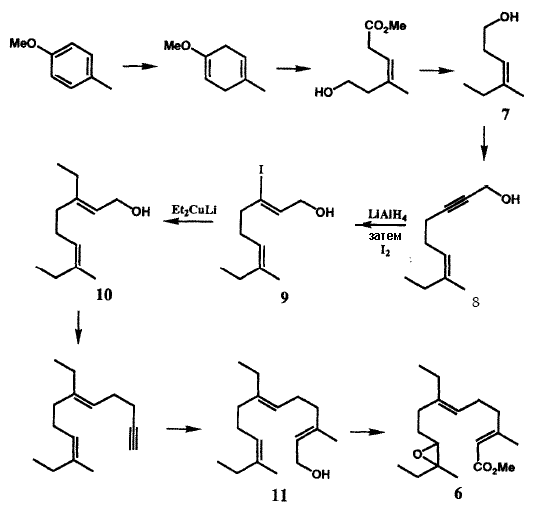
Легкодоступность ювенильного гормона насекомых 6 позволила провести широкий спектр биологических исследований и достичь понимания наилучших путей использования таких препаратов «третьего поколения» для борьбы с насекомыми. Недорогой синтетический миметик ювенильного гормона насекомых в настоящее время производится в коммерческих масштабах как экологически безопасное средство борьбы с насекомыми.
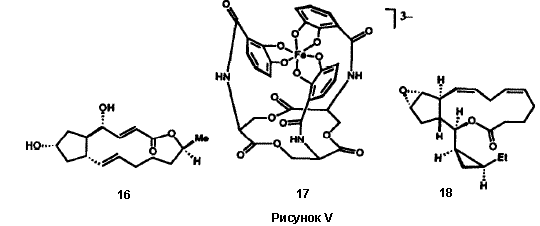
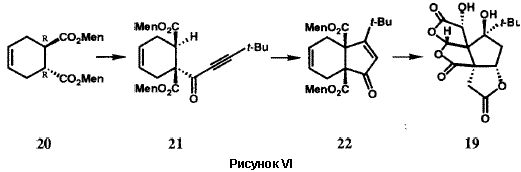
Многочисленные встречающиеся в природе вещества микробного происхождения, в особенности антибиотики, являются членами структурного ряда «макролидов» и содержат лактонную функциональную группу как часть многочленного цикла группу. Ключом к успешному синтезу сложных макролидов, таких как эритронолид B (12), прекурсор эритромициновых антибиотиков, была разработка новой методологии образования макролактонного кольца (Рис. V). Наша группа разработала очень мягкий, эффективный и общий метод для этой синтетической операции, метод двойной активации 19, который впоследствии стал широко использоваться. Тиоэфир 13, получаемый при полном синтезе 20, превращался в макролактон 15 простым нагреванием в толуоле. Внутримолекулярный перенос протона в 13 генерирует внутримолекулярную ионную пару 14, которая дважды активирована для внутримолекулярного присоединения по карбонильной группе, необходимого для замыкания цикла с образованием 15. Эритронолид B был получен из 15 в результате простой последовательности реакций.20 Метод двойной активации был с успехом использован для синтеза большого числа других примечательных природных макроциклических лактонов (Рисунок V), включая брефельдин A (16), ингибитор транспорта и переработки белков в клетках млекопитающих,21 переносчик железа в микроорганизмах - энтеробактин (17),22 и эйкозаноид морского происхождения, гибридалактон (18).23 Билобалид (19) является сложной и необычной молекулой, продуцируемой деревом гинкго, Ginkgo biloba. Успешный синтез 19 стал возможен в результате разработки примечательной реакции для образования пятичленных циклов (Рисунок VI).24 Легкодоступный хиральный диэфир 20 был преовращен в результате реакции ацилирования Кляйзена в ацетиленовый кетодиэфир 21. Обработка 21 основанием вызвала замыкание нового цикла с образованием бициклического кетона 22, который затем был превращен в билобалид (19) в результате многостадийной последовательности реакций. Имеется несколько вариантов такой методологии циклизации, которая демонстрирует значительные возможности 25. В новой методологии, разработанной в нашей группе, большую роль играют широкий диапазон реагентов и реактантов: переходные металлы; комплексы ионов металов; а также кремний-, сульфо-, боро-, алюмино-, фосфо- и станно-замещенные карбогены. Созданные в результате новые общие методы включают в себя процессы образования циклов, удлинения цепи, окисления, восстановления, преобразования функциональных групп, их активирования, защиты и управления стереохимией.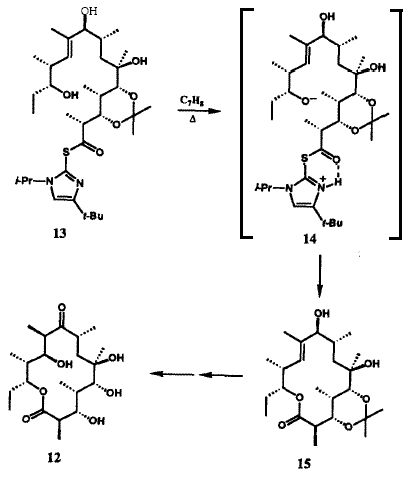
Многостадийный синтез - общие вопросы
Для химика-синтетика сложные молекулы природного происхождения так же привлекательны как и молекулы любого другого происхождения. Восприятие этой привлекательности зависит от понимания химического строения и их трансформаций, и, точно так же, как и в случае произведения искусства, углубляется по мере изучения предмета, возможно приближаясь даже к уровню романса. Не удивительно, что современный химик-синтетик наполняется радостью при открытии новой природной структуры или появлении еще одной сложной синтетической задачи. Нет разницы, что реализация сложного синтеза влечет за собой долгие часы исследований, умственного и физического напряжения, поскольку сложный химический синтез является волнующим приключением, которое ведет к красивому творению. Я верю в то, что аргументы в пользу молекулярного синтеза, как высокоинтеллектуальная попытка и как произведение научного искусства, can stand on these merits. Химик, который разрабатывает и выполняет оригинальный и эстетически привлекательный многостадийный синтез подобен композитору, художнику или поэту, который с высокой степенью индивидуальности моделирует новые виды красоты из взаимодействия разума и духа. Весьма удачно, что молекулярный синтез служит также утилитарным потребностям производства определенных количеств редких или новых веществ, которые удовлетворяют человеческие потребности, в особенности качающихся здоровья, а также научную функцию стимулирования исследований и образования во всей науке химии. Наша научная группа ответственна за создание более чем сотни новых многостадийных синтезов интересных молекул, наших сонат и струнных квартетов. Пошаговое конструирование большинства этих ецлевых продуктов синтеза описано в книге «Логика химического синтеза»7 и подробно обсуждается в оригинальных статьях, цитируемых в этой книге. Структуры небольшой и отчасти случайной подборки этих целевых молекул показаны на рисунках VII a и VII b. Здесь мы приводим небольшие комментарии по синтезу каждой из них, чтобы дать обзор этого аспекта этого аспекта наших исследований. Мейтанзин 26a и аплазмомицин,26b each scarce и терапевтически очень интересные вещества, были энантиоселективно синтезированы с управлением стереохимией с использованием новой методологии для сборки скелета молекулы и формирования макроциклического фрагмента. Эти синтезы и синтез эритронолида B19 6c дают первый пример того, что такие сложные, макроциклические молекулы могут быть эффективно получены с помощью полного многостадийного синтеза.
Гиббереллиновая кислота не поддавалась полному синтезу, несмотря на изучение в различных ведущих лабораториях, в течение более чем двух десятилетий вследствие необычно неприступного расположения структурных фрагментов. Первый удачный синтез и последующие его усовершенствованные версии, 26d потребовали глубокого и комплексного ретросинтетического анализа 26e, а также большого числа новых концепций и методов. Совершенно другой стратегический подход был использован для первого синтеза биосинтетически родственного регулятору роста растений - антеридиновой кислоты 26f, которая бы соответствовала предполагаемой большой структуре и проясняла стереохимию. Доступность синтетической антеридиновой кислоты весьма существенно для дальнейшего изучения этого редкого и мощного растительного гормона.
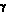 -аминомасляной кислоты, был бы невозможен без ретросинтетического анализа и новой методологии.26h
-аминомасляной кислоты, был бы невозможен без ретросинтетического анализа и новой методологии.26h 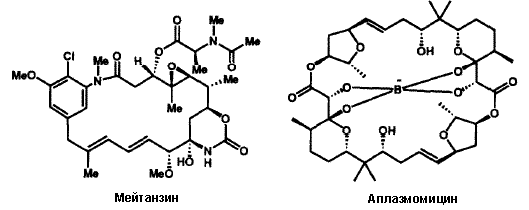
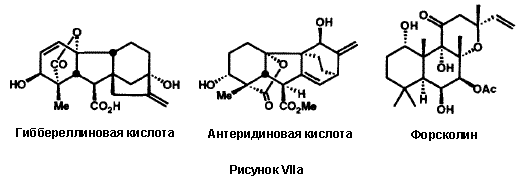
Пергидрогистрионикотоксин, редкий и обладающий высокой биологической активностью алкалоид из яда лягушек, являющийся полезным инструментом в неврологии, был без труда получен в результате полного синтеза.26i
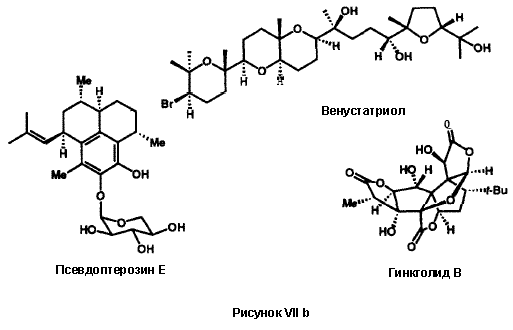
Псевдоптерозин E26j, мощный противовоспалительный препарат, и венустатриол26k, антивирусный препарат, биосинтезируются морскими организмами только в следовых количествах. Оба они стали доступными в хиральной форме в результате энантиоконтролируемого многостадийного синтеза.
Последним, но не последним по значению, в приведенном списке наших исследований в области полного синтеза сложных молекул, является случай гинкголида B, необычного во многих отношениях вещества. Гинкголид B биосинтезируется в корнях уникального и ископаемого дерева гинкго, Ginkgo biloba, в результате чрезвычайно сложного биосинтетического процесса, по причинам, которые неизвестны. Он является активным ингредиентом в медицинском экстракте ginkgo. который в настоящее время широко используется в восточной и западной медицине. Полный синтез гинкголида B являля очень сложной задачей для синтеза в 1980-е годы, которая была сравнима с наиболее сложными задачами более ранних эпох, например, синтеза стероидов в 1950-е годы или витамина B-12 и гиббереллиновой кислоты в 1970-е. Этот задача была решена только в результате трех лет исследований, вновь благодаря мощи современного ретросинтетического планирования и разработке новых инструментов для этого частного синтеза.26l,27
Примеры многостадийных синтезов - простагландины
Простагландины, первые из известных эйкозаноидов, были обнаружены как биоактивные вещества более чем 50 лет тому назад. Однако до пионерских работ К. Суна Бергстрема и его группы в Швеции в 1950-е и 1960-е г.г. строение этих различных членов семейства простагландинов определено не было.28 За это исследование Бергстрем и Бегт Самуэльсон получили (совместно с Джоном Вэйном) Нобелевскую премию по медицине за 1982 г., и вполне заслуженно, поскольку как было написано о простагландинах: «Их действие и фармакологические агенты, которые влияют на их образование оказывают влияние практически на каждый аспект медицинской практики».29 Присутствие лишь следовых количеств простагландинов (PG) в тканях млекопитающих и мощное влияние этих двенадцати-углеродных карбоксильных соединений на мускулы и кровеносные сосуды свидетельствует о необходимости создания эффективного метода синтеза, что сделает доступными все PG в количествах, достаточных для изучения исследования их физиологического действия и терапевтического использования. Задача химического синтеза была сложной вследствие неопределенностей, касающихся стабильности и химии PG, а также вследствие существования трех различных семейств (PG1, PG2 и PG3), каждое из которых состоит из нескольких членов. Первый полный синтез основных PG, опубликованный в 1967,30,31 сделал доступным важные члены первого семейства (PGA1, PGE1 и PGF1a) и позволил оценить их химические свойства, которые облегчили создание общего метода синтеза всех этих простагландинов. Общий синтез простагландинов предоставляет доступ ко всем PG из одного единственного интермедиата, обычно известного как лактон-альдегид Кори. 32,33 В различных формах эти гибкие синтезы были использованы лабораториями всего мира для получения не только природных PG, но также и бессчетных структурных аналогов в любых количествах.34
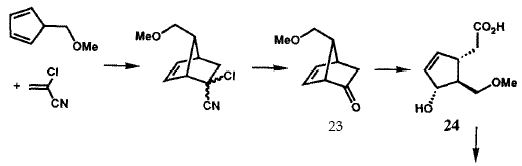
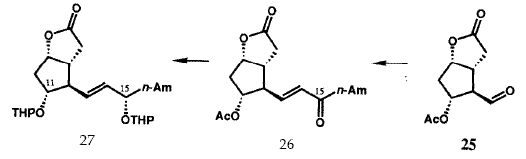
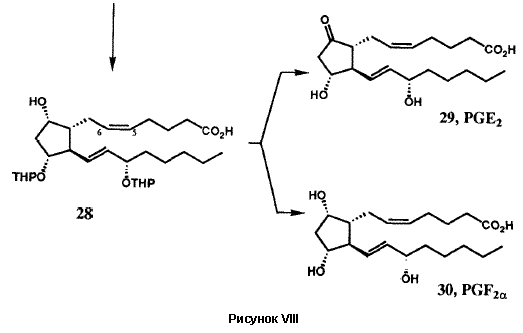
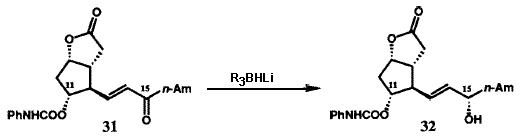
Исходная версия проведенного в 1969 г. общего синтеза PG в общих чертах представлена на рисунке VIII. Бициклогептенон 23 был стереоспецифично синтезирован с помощью новой катализируемой ионами Cu(II) реакции Дильса-Альдера с последующим щелочным гидролизом конечного аддукта. Пероксиды в щелочной среде превращают 23 в гидроксикислоту 24, которая легко может быть расщеплена с использованием (+)-эфедрина. Лактонизация и операции по замене функциональных групп преобразуют 24 в лактон-альдегид Кори 25, универсальный прекурсор всех PG и их аналогов. Енон 26 (Am = C5H11), стереоспецифически получаемый из 25 с помощью реакции сочетания Хорнера-Эммонса, при восстановлении боргидридом цинка дает требуемый 15-(S)-спирт наряду с 15-(R)-диастереомером, который был отделен и утилизирован через 26 в 15-(S)-спирт. Защита гидроксильных групп у C(11) и C(15) осуществлялась получением соответствующего бистетрагидропиранильного (bis THP) эфира, 27. Восстановление лактонной функции 27 в лактол (с помощью R2AIH) и сочетание Виттига стереоспецифично давало 28. Кислотный гидролиз 28 давал PGF2a (30), тогда как окисление 28 с последующим гидролизом приводило к PGE2(29). Гидрирование 5,6-двойной связи в 28 с последующими теми же самыми оконечными стадиями дает PGF1a и PGE1. Парпллельные серии трансформаций были использованы для превращения 25 в PGE3a и PGE3. Хотя разработанный в 1969 бисциклогептеноновый путь синтеза PG был высокоэффективен для синтеза в больших масштабах, он не был окончательным. Путь через реакцию Дильса-Альдера к 23 дает рацемический продукт, что в свою очередь влечет за собой необходимость расщепления гидроксикислоты 24. Другой проблемой было отсутствие стереоспецифичности в реакции восстановления кето-группы при C(15) соединения 26. Оба эти ограничения были преодолены в результате изобретения новой методологии, которая одновременно открыла большие новые области синтетических проектов.
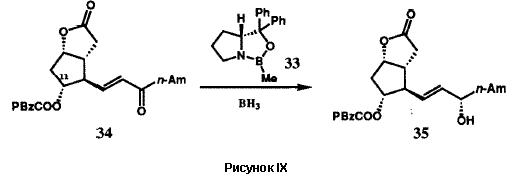
Оксазаборолидин 33 является замечательным каталитическим реагентом. Он управляет абсолютной стереохимией реакции восстановления самых разнообразных кетонов дополнительно к тому, что он еще и ускоряет реакцию восстановления бораном. Абсолютная стереохимия продукта восстановления ахирального кетона RSCORL, где RS меньше, чем RL, предсказуема.37,38 Наблюдаемый катализ и энантиоселективность согласуются с механизмом, представленным на рисунке X. Катализатор 33, как показано, стереоспецифично образует комплекс с бораном с образованием частиц, которые активированы для присоединения к кетонному субстрату. Комплексообразование по стерически более доступной неподеленной электронной пары карбонильного атома кислорода и внутримолекулярный перенос гидрид-иона приводят к наблюдаемому энантиомеру вторичного спирта. В этом механизме реагент 33 фактически действует как молекулярный робот: он вначале захватывает и удерживает один из реактантов, BH3, и становится более активным в отношении другого реактанта. Затем он присоединяется к кетону с образованием строго определенного трехмерного молекулярного ансамбля, который способствует переносу водорода между двумя реактантами с образованием определенного энантиомера продукта восстаановления. Этот молекулярный робот в конечном итоге выгружает образовавшиеся продукты и повторяет реакционный цикл. Такое действие небольшой молекулы 33 напоминает катализ ферментами, которые также могут рассматриваться как молекулярные роботы. Естественно, у 33 отсутствует функция дифференциации субстратов по форме и размеру, как у ферментов, поскольку он слишком мал, чтобы обладать связывающим карманом или периферийным мультиконтактным центром распознавания.
Действие молекулярного робота 33 представляет собой новое направление химического синтеза. В прошлом, большинство синтетических конструкций зависели от парных столкновений между реактантами без содействия со стороны молекулярных роботоподобных сборщиков. Вполне вероятно, что синтетическая химия в будущем даст нам много новых молекулярных роботов, в качестве части своего прогресса на пути ко все более большим высотам.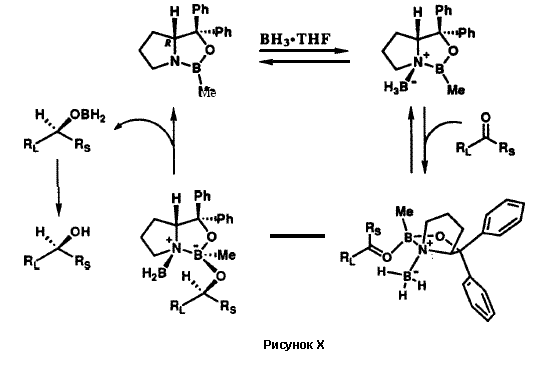
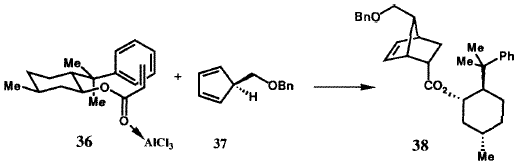
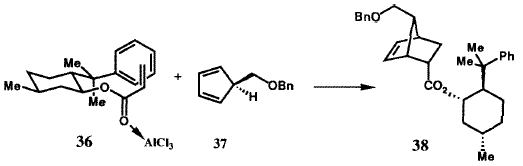
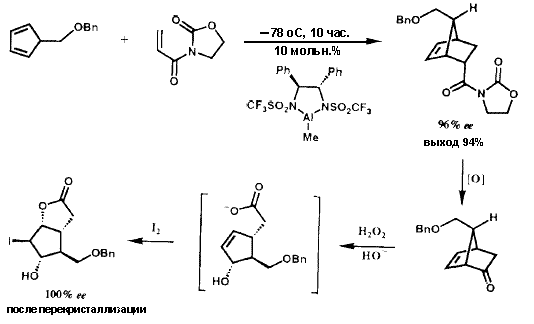
Были разработаны два решения для энентиоселективного синтеза бициклогептенона 23 (рисунок VIII) с необходимой хиральностью для получения природных простагландинов. В первом из них была использована стереоокнтролируемая катализируемая хлоридом алюминия реакция Дильса-Альдера между бензилоксиметилциклопентадиеном (37) (рисунок XI) и акриловым эфиром S-фенилментола (36), в результате которой требуемый аддукт 38 образовался с очень высокой (32: 1) энантиоселективностью.39 Аддукт 38 через кетон 23 был преобразован в иодолактон 39, который был получен в энантиомерно чистом виде с высоким выходом в результате простой перекристаллизации и превращен в стандартный интермедиат PG 40. Кроме того, 8-фенилментол был эффективно возвращен. Энантиоселективное образование 38 может быть понято из геометрии, показанной для комплекса 36·A1C13, и стерического отбора, осуществляемого фенильной группой, с si (тыльной) стороны акрилатной  двойной связи. Эффективность S-фенилментольного контроллера стимулировало развитие других контроллеров (хиральных вспомогательных групп) для использования в энантиоселективном синтезе.40 Совсем недавно, это достижение было превойзодено разработкой молекулярного робота, который собирает ахиральные компоненты, как это показано на рисунке XII, с образованием необходимого аддукта реакции Дильса-Альдера с выходом 94 % и почти 50:1 энантиоселективностью. 38,41 Превращение энантиомерно чистого иодолактона 39 было осуществлено с использованием стандартных методик.42
двойной связи. Эффективность S-фенилментольного контроллера стимулировало развитие других контроллеров (хиральных вспомогательных групп) для использования в энантиоселективном синтезе.40 Совсем недавно, это достижение было превойзодено разработкой молекулярного робота, который собирает ахиральные компоненты, как это показано на рисунке XII, с образованием необходимого аддукта реакции Дильса-Альдера с выходом 94 % и почти 50:1 энантиоселективностью. 38,41 Превращение энантиомерно чистого иодолактона 39 было осуществлено с использованием стандартных методик.42
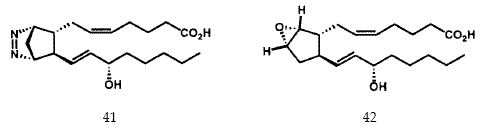
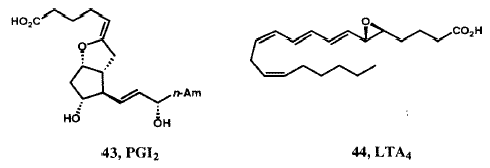
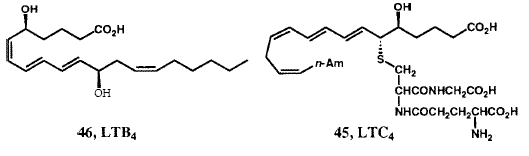
Биосинтез простагландинов происходит в результате окислительного превращения C20-ненасыщенной кислоты - арахидоновой кислоты с образованием бициклического эндопероксида PGH2, который служит прекурсором не только для PGE2 и PGF2a, но и PGI2 и тромбоксана A2.43 Нам удалось синтезировать стабильный активный азо-аналог (41) неустойчивого PGH2 44 (Рисунок XIII) и стабильные активные аналоги (например, 42) нестабильного тромбоксана A2, 45, а также самого PGI2 (43).46 В 1977 мы предложили структуру нестабильного эйкозаноида, который известен как лейкотриен A4 (LTA4) (44) и, в начале 1979 г., синтезировали эту структуру в предвкушении его выделения из природных источников. 47 В результате совместных исследований нашей группы и команды Karolinska, возглавляемой Бенгтом Самюэльсоном, было установлено, что LTA4 соединяется с глютатионом с первоначальным образованием «медленно реагирующей субстанции», в настоящее время известной как LTC4 (45).47 Подробная стереохимия хемотактического лейкотриена LTB4 (46) была вначале установлена в результате синтеза. 47,48 Химический синтез этого лейкотриена сделал эти соединения доступными в количествах, необходимых для многих сотен биологических исследований, которые и были проведены впоследствии. Новые полезные соединения, которые проявляют активность в качестве антагонистов PGF2a, 49 тромбоксана A2, 50 и LTB4,51 и как ингибиторы биосинтеза лейкотриена 52 также появились в ходе выполнения нашей программы.
Я надеюсь, что наши исследования в области эйкозаноидов 34 окажется предвестником будущих академических синтетических проектов, поскольку в настоящее время имеется беспримерная возможность для применения химического синтеза к биологическим и медицинским проблемам на фундаментальном уровне.
Благодарности
Мне доставляет большое удовольствие выразить мою глубокую благодарность многим личностям, кто deserve credit for the accomplishments summarized herein: моим учителям в Lawrence High School и Массачусетском Технологическом Институте, моим коллегам в Университете Иллинойса и Гарвардском Университете, моим замечательным научным сотрудникам и моей дорогой семье. Исследовательские гранты от National Science Foundation, the National Institutes of Health, Pfizer Inc. и многочисленных других доноров обеспечили необходимую финансовую поддержку.
СПИСОК ЛИТЕРАТУРЫ
1. N. Anand, J. S. Bindra, and S. Ranganathan, Art in Organic Synthesis (Holden-Day, Inc., San Francisco, first edition, 1970). Эта книга содержит обзор этих и других примечательных синтезов периода 1940 - 1970 гг.2. R. B. Woodward, Les Prix Nobel en 1965, (Almquist and Wiksell, Intl., Stockholm, 1966) p. 192.
3. E. J. Corey, Pure and Applied Chem., 14, 19 (1967).
4. E. J. Corey and W. T. Wipke, Science, 166, 178 (1969).
5. E. J. Corey, Quart. Rev. Chem. Soc., 25, 455 (1971).
6. (a) E. J. Corey, M. Ohno, P. A. Vatakencherry, and R. B. Mitra Am. Chem. Soc., 83, 1251 (1961); (b) те же J. Am. Chem. Soc., 86, 478 (1964).
7. E. J. Corey and X.-M. Cheng, The Logic of Chemical Synthesis (John Wiley and Sons, Inc., New York, 1989).
8. Этот раздел взят в основном из ссылки 7.
9. E. J. Corey, R. D. Cramer, III, and W. J. Howe, J. Am. Chem. Soc ., 94, 440 (1972).
10. Ссылка 7, Часть 6.
11. E. J. Corey, A. K. Long, and S. D. Rubenstein, Science , 228, 408 (1985).
12. E. J. Corey, W. T. Wipke, R. D. Cramer, III, and W. J. Howe, J. Am. Chem. Soc., 94, 421 (1972).
13. Этот компьютерный анализ был осуществлен Джоном Каппосом в нашей группе LHASA.
14. H. Roller, K.-H. Dahm, C. C. Sweeley, and B. M. Trost, Angew. Chem. Int. Ed. Engl., 6, 179 (1967).
15. S. S. Tobe and B. Stay, Adv. Insect. Physiol., 18, 305 (1985).
16. (a) E. J. Corey, J. A. Katzenellenbogen, N. W. Gilman, S. A. Roman, and B. W. Erickson, J. Am. Chem. Soc., 90, 5618 (1968); (b) E. J. Corey, Bull. Soc. Ent. Suisse, 44, 87 (1971); (c) ссылка 7, p. 146; (d) E. J. Corey and G. H. Posner, J. Am. Chem. Soc., 89, 3911 (1967).
17. G. H. Posner, An Introduction to Organic Synthesis Using Organocopper Reagents (John Wiley and Sons, Inc., New York, 1980).
18. E. J. Corey and J. A. Katzenellenbogen, J. Am. Chem. Soc., 91, 1851 (1969). 19. (a) E. J. Corey and K. C. Nicolaou, J. Am. Chem. Soc., 96, 5614 (1974); (b) E. J. Corey, K. C. Nicolaou, and L. S. Melvin, Jr., J. Am. Chem. Soc., 97, 654 (1975); (c) E. J. Corey, D. J. Brunelle, and P. J. Stork, Tetrahedron Lett., 3405 (1976); (d) E. J. Corey and D. J. Brunelle, Tetrahedron Lett., 3409 (1976).
20. E. J. Corey, S. Kim, S.-e. Yoo, K. C. Nicolaou, L. S. Melvin, Jr., D.J. Brunelle, J. R. Falck, E. J. Trybulski, R. Lett, and P. W. Sheldrake, J. Am. Chem. Soc., 100, 4620 (1978).
21. (a) E. J.Corey and R. H. Wollenberg, Tetrahedron Lett., 4705 (1976); (b) E. J. Corey, R. H. Wollenberg, and D. R. Williams, Tetrahedron Lett., 2243 (1977); E. J. Corey and P. Carpino, Tetrahedron Lett., 31, 7555, 1990.
22. E. J. Corey and S. Bhattacharyya, Tetrahedron Lett., 3919 (1977).
23. E. J. Corey and B. De, J. Am. Chem. Soc., 106, 2735 (1984).
24. (a) E. J. Corey and W.-g. Su, J. Am. Chem. Soc., 109, 7534 (1987); (b) E. J. Corey and W.-g. Su, Tetrahedron Lett., 29, 3423 (1988).
25. (a) E. J. Corey, W.-g. Su, and I. N. Houpis, Tetrahedron Lett., 27, 5951 (1986); (b) E. J. Corey and W.-g. Su, Tetrahedron Lett., 28 , 5241 (1987).
26. (a) .Ссылка 7, pp. 116-123; (b) ссылка 7, pp. 128-133; (c) ссылка 7, pp. 104-107; (d) ссылка 7, pp. 205-211; (e) ссылка 7, pp. 84-85; (f) ссылка 7, pp. 212-214; (g) ссылка 7, pp. 230-233; (h) ссылка 7, pp. 86-87, 178-179; (i) ссылка 7, pp. 83-84, 136-137; (j) ссылка 7, pp. 237-238; (k) ссылка 7, pp. 234-26; (1) ссылка 7, pp. 89-91; 221-226.
27. E. J. Corey, Chem. Soc. Rev., 17, 111 (1988).
28. S. Bergstrom, Science, 157, 382 (1967).
29. J. A. Oats, G. A. Fitzgerald, R. A. Branch, E. K. Jackson, H. R. Knapp, and L. J. Roberts, II, New Eng. J. Med. , 319, 689 (1988).
30. (a) E. J. Corey, N. H. Andersen, R. M. Carlson, J. Paust, E. Vedejs, I. Vlattas, and R. E. K. Winter, J. Am. Chem. Soc., 90, 3245 (1968); (b) E. J. Corey, I. Vlattas, N. H. Andersen, and K. Harding, J. Am. Chem. SOc, 90,3247 (1968); (c) E. J. Corey, I. Vlattas, and K. Harding, J. Am. Chem. Soc., 91, 235 (1969); (d) E. J. Corey, Ann. New York Acad. Sci., 180, 24 (1971).
31. Ссылка 7, стр. 250-254.
32. (a) E. J. Corey, N. M. Weinshenker, T. K. Schaaf, and W. Huber, J. Am. Chem. Soc., 91, 5675 (1969); (b) E. J. Corey, T. K. Schaaf, W. Huber, U. Koelliker, and N. M. Weinshenker, J. Am. Chem. Soc., 92,397 (1970); (c) E. J. Corey, R. Noyori, and T. K. Schaaf, J. Am. Chem. Soc., 92, 2586 (1970).
33. Ссылка 7, pp. 255-296.
34. В качестве другого общего отчета по этому проекту и последующих исследованиях эйкозаноидов, см. E. J. Corey, Japan Prize in Science for 1989, Annual Report of the Science and Technology Foundation of Japan, pp. 95 - 109, 1989.
35. E. J. Corey, S. M. Albonico, U. Koelliker, T. K. Schaaf, and R. K. Varma, J. Am. Chem. Soc. , 93, 1491 (1971).
36. E. J. Corey, R. K. Bakshi, S. Shibata, C.-P. Chen, and V. K. Singh, J. Am. Chem. Soc., 109, 7925 (1987).
37. E. J. Corey, R. K. Bakshi, and S. Shibata, J. Am. Chem. Soc., 109, 555, (1987).
38. E. J. Corey, Pure and Appl. Chem., 62, 1209 (1990).
39. E. J. Corey and H. E. Ensley, J. Am. Chem. Soc., 97, 6908 (1975).
40. (a) W. Oppolzer, Angew. Chem. Int. Ed. Engl., 23, 876 (1984); (b) G. Helmchen, R. Karge, and J. Weetman, Modern Synthetic Methods Vol. 4 (Springer-Verlag, Berlin, 1986), R. Scheffold, ed., p. 261.
41. E. J. Corey, R. Imwinkelried, S. Pikul, and Y. B. Xiang, J. Am. Chem. Soc., 111, 5493 (1989).
42. E. J. Corey and N. Imai, Tetrahedron Lett , в печати (1991).
43. N. A. Nelson, R. C. Kelly, and R. A. Johnson, Chem. Eng. News, 60, 30 (1982).
44. (a) E. J. Corey, K. C. Nicolaou, Y. Machida, C. L. Malmsten, and B. Samuelsson, Proc. Nut. Acad. Sci. USA, 72, 3355 (1975); (b) E. J. Corey, K. Narasaka, and M. Shibasaki, J. Am. Chem. Soc., 98, 6417 (1976).
45. (a) T. K. Schaaf, D. L. Bussolotti, M. J. Parry, and E. J. Corey, J. Am. Chem. Soc., 103, 6502 (1981); (b) E. J. Corey and W.-g. Su, Tetrahedron Lett., 31, 2677 (1990).
46. E. J. Corey, G. E. Keck, and I. Szekely, J. Am. Chem. Soc., 99, 2006 (1977); (b) E. J. Corey, H. L. Pearce, I. Szekely, and M. Ishiguro, Tetrahedron Lett., 1023 (1978).
47. (a) E. J. Corey, Experientia, 38, 1259 (1982); (b) ссылка 7, pp. 312-317; (c) E. J. Corey, Y. Arai, and C. Mioskowski, J. Am. Chem. Soc., 101,6748 (1979); (d) E. J. Corey, D. A. Clark, G. Goto, A. Marfat, C. Mioskowski, B. Samuelsson, S. Hammarstrom, J. Am. Chem. Soc., 102, 1436, 3663 (1980).
48. (a) E. J. Corey, A. Marfat, G. Goto, and F. Brion, J. Am. Chem. Sac., 102, 7984 (1980); (b) E. J. Corey, A. Marfat, J. E. Munroe, K. S. Kim, P.B. Hopkins, and F. Brion, Tetrahedron Lett., 22, 1077 (1981).
49. R. B. Stinger, T. M. Fitzpatrick, E. J. Corey, P. W. Ramwell, J. C. Rose, and P. A. Kot, J. Pharm. Exp. Ther., 220, 521 (1982).
50. E. J. Corey and W.-g. Su, Tetrahedron Lett., 31, 3833 (1990).
51. H. J. Showell, I. G. Otterness, A. Marfat, and E. J. Corey, Biochem. Biophys. Res. Commun., 106, 741 (1982). 52. ссылка 7, стр. 345 - 352.